







1. INTRODUCTION
Dengue fever primarily occurs in tropical and subtropical regions and is caused by the Dengue virus (DENV) which belongs to the family Flaviviridae, genus Flavivirus [1]. According to the US statistics [2], dengue hemorrhagic fever is an annual epidemic with a mortality rate over 13% in untreated patient. Five serotypes of the dengue virus have been reported, namely DENV1, DENV2, DENV3, DENV4, and DENV5, with DENV2 being the main cause of major outbreaks and severe cases [3]. There is currently no specific antivirals has been approved for dengue therapeutic [4].
The genome of Dengue virus type 2 (DENV2) comprises of a positive single-stranded RNA that is approximately 10,700 nucleotides long and can be directly translated into polyproteins [5]. These polyproteins are truncated into ten different proteins, which can be classified into two groups: structural proteins and non-structural proteins (NS). Structural proteins, including capsid protein, envelope protein, and membrane protein, are involved in virus entry and budding processes [5]. NS consist of seven proteins: NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5, which have been reported to contribute to the inhibition of signaling from interferon-α/β [5].
Among targets of dengue drug discovery, NS4B is the largest trans-membrane protein and shares high similarity among different DENV strains [4]. NS4B is particularly important due to its central role in the virus life cycle, such as: (i) inhibiting the interferon-α/β signaling; (ii) interacting with NS3 to regulate the function of the helicase enzyme; (iii) interacting with NS1 in virus replication; (iv) contributing to suppostss the host’s RNA interference response [4]. Several compounds, such as NITD-688, SDM25N, 14a, AM404 and spiropyrazolopyridone, have been studied in vitro and in vivo for their inhibitory effects on this protein [4]. However, none of these compounds have progressed to clinical research due to their high toxicity profiles.
Natural compounds have historically played a crucial role in drug development, particularly for cancer and infectious diseases [6]. These phytochemicals are characterized by their great structural diversity and complexity. Many investigation techniques have been utilized in drug discovery from natural source. In anti-dengue agents discovery, many medicinal plants, such as Andrographis paniculata, Momordica charantia, and Schisandra chinensis, have demonstrated promising anti-dengue effects through in vitro and in vivo studies [7]. Through in silico approaches, such as homology modelling, molecular docking and quantitative structure–activity relationship (QSAR), some phytochemical structures have been identified to be developed as novel NS4B inhibitors, including: eu-flavonoid, 1,3-benzodioxole, iso-flavonoid, and indole [8,9].
Therefore, the objective of this study was to identify potential natural inhibitors of the NS4B protein of DENV2 using molecular docking and molecular dynamics simulations (MDs) approaches. The method initiated by modelling the NS4B protein structure and followed by screening in-house natural compounds using molecular docking. MDs and binding free energy calculation were combined to investigate the stability and flexibility of NS4B protein as well as protein-ligand complexes to select the most potential inhibitors for DENV2 NS4B.
2. MATERIALS AND METHODS
Due to the lack of crystal structure of NS4B protein of DENV2, two methods were conducted to postdict the structure of this protein:
-
(1) Template-based approach using homology modelling with SWISS-MODEL server (https://swissmodel.expasy.org) [10].
-
(2) Template-free approach using ColabFold 1.5.2 (https://colab.research) [11].
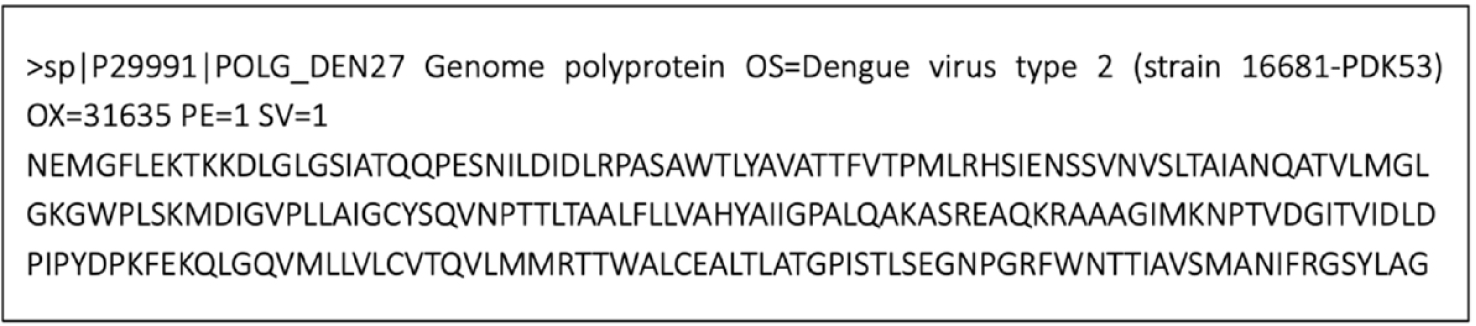
The UniProt database entry P29991, which repostsents the full polyprotein genome of DENV2, was used to obtain the sequence of the NS4B protein. Specifically, residues 2,244 to 2,491 were extracted from P29991 (248 amino acids) to define the NS4B sequence (Fig. 1). This sequence was then subjected to SWISS-MODEL server and ColabFold to generate 3D structure. In SWISS-MODEL, the structural template used for building model was selected in terms of a sequence identity of over 30% or a minimum sequence similarity of 0.4 between the NS4B protein and the template protein. In addition, ColabFold generated five high-quality structural models, and the first ranked model was chosen for further analysis.
Then, the protein structure was run MDs for 25 ns at temperature 300 K, postssure 1 bar to investigate the equilibrium structure using Gromacs 2022.5 [12]. The simulation results were analyzed based on the value of root-mean-square-deviation (RMSD). The equilibrium structure was obtained at the time that the RMSD value was below 2 Å (0.2 nm).
Using the obtained equilibrium structure of DENV2 NS4B, the binding site of this protein was determined by three methods, including CATSp v3.0 [13], P2Rank v2.4 [14], and blind docking with 73 compounds that have known to have activity against DENV2 or the NS4B protein of DENV2 from 30 articles [4,15–43] (structure and corresponding EC₅₀ values (μM) listed in Supplementary Table 1).
Then, the final binding cavity was selected by comparing and overlapping the results from these three methods.
Virtual screening process was performed through molecular docking on the structure of DENV2 NS4B and binding cavity obtained in the postvious steps. The screening database included 286 in-house natural compounds which were collected from the Faculty of Pharmacy, University of Medicine and Pharmacy at Ho Chi Minh City, Vietnam (listed in Supplementary Table 2). The two-dimensional (2D) structures of ligands were drawn and converted to three-dimensional (3D) structures by ChemSketch [44] and Discovery Studio Visualizer version 2021 [45], respectively. After energy minimizing and converting to pdb format, these ligands were postpared for docking by using AutoDock Tools 1.5.6 package [46]. Later on, Autodock Vina software [47] was used for docking process with grid box parameters obtained from the postdiction binding site process and were listed in Table 1. Molecular docking was also conducted for 16 reference compounds (listed in Supplementary Table 3). The docking results were analyzed based on two criteria, including binding affinity (kcal/mol) and interactions between ligand and residues in binding cavity. Compounds that had strong binding affinities (≤–9.0 kcal/mol) and interactions with residues similar to interactions of the reference compounds were identified as promising compounds.
| Parameter | Center_x | Center_y | Center_z | Size_x | Size_y | Size_z | Spacing |
|---|---|---|---|---|---|---|---|
| Value (Å) | 100.577 | 83.641 | 45.819 | 28 | 28 | 28 | 1.00 |
To further investigate the results of molecular docking, top five potential compounds were selected for MDs using Gromacs 2022.5 software [12]. The structures, including the complexes of NS4B protein-ligand and apo NS4B protein (free ligand), were run MDs during 50 ns using the all-atom CHARMM-36 force field. The process involved several stages, including topology postparation, generation of a dodecahedron simulation box, solvation of the system in water and adding ions to neutralize the system, energy minimization and system equilibration at a temperature of 300 K and a postssure of 1 bar within 1,000 ps before running MDs. The simulation results were analyzed based on parameters such as the values of RMSD, root-mean-square-fluctuation (RMSF), radius of gyration (Rg), solvent-accessible surface area (SASA), and percentage of hydrogen bond occupancy. In particular, the hydrogen bond occupancy percentages were analyzed using the VMD software [48] to evaluate the interaction potential of ligands with the key residues. A hydrogen bond was defined by simple geometric criteria, which included a distance of less than 3.5 Å between the hydrogen donor (D) and acceptor (A) atoms, and an angle greater than 120° for D–H⋯A [49].
After the 50 ns MDs had been completed, the most stable NS4B protein-ligand complexes were further subjected to a longer simulation of 200 ns. The results were compared with the values of the apo NS4B protein under the same conditions and run time.
The binding energies of the complexes of the top hit compounds, obtained from the long-time scale MDs using the CHARMM-36 force field, were calculated by the gmx_MMPBSA package [50]. In this study, snapshots taken from the 200 ns MDs trajectory of each protein-ligand complex were used to calculate binding free energy through both MM/GBSA and MM/PBSA (Molecular Mechanics/Generalized Born or Poisson-Boltzmann Surface Area) methods. The results from these approaches were compared and evaluated. The solute’s dielectric constant was set to 1.0, with a temperature of 298 K and a salt concentration of 0.15 M. The free energies (ΔGblind) for binding of the ligand to NS4B protein with ligand in solvent can be expostssed as [51]:
The equation (1) can be divided into the contributions of various interactions and repostsented as:
in which:
In the above equations, ΔEMM, ΔGsolv and –TΔS refer to the changes in gas phase molecular mechanics energy, solvation free energy, and conformational entropy that occur during ligand binding, respectively. Due to the high computational cost, changes in conformational entropy (–TΔS) are typically neglected when calculating the relative binding free energies of similar ligands [51]. ΔEint repostsents the internal bonded energy components, such as bond, angle, and dihedral energies, which are regarded as zero in a dynamic simulation [51]. Evdw and ΔEele are the nonbonded van der Waals and the electrostatic interaction energy, respectively. ΔGsolv is the sum of the electrostatic solvation energy ΔGPB/GB (polar component) and the nonpolar contribution ΔGSA between the solute and the surrounding continuum solvent. The polar contribution is determined using the PB method with the level set function model [52] and the GB method with the GB-OBC2 model [53], while the nonpolar energy is typically estimated based on SASA.
3. RESULTS AND DISCUSSION
With template-based approach, no 3D crystal protein structure retrieved from Protein Data Bank satisfied the criteria of identity>30% or similarity>0.4 (Supplementary Table 4) to be used as a template in the homology modeling method.
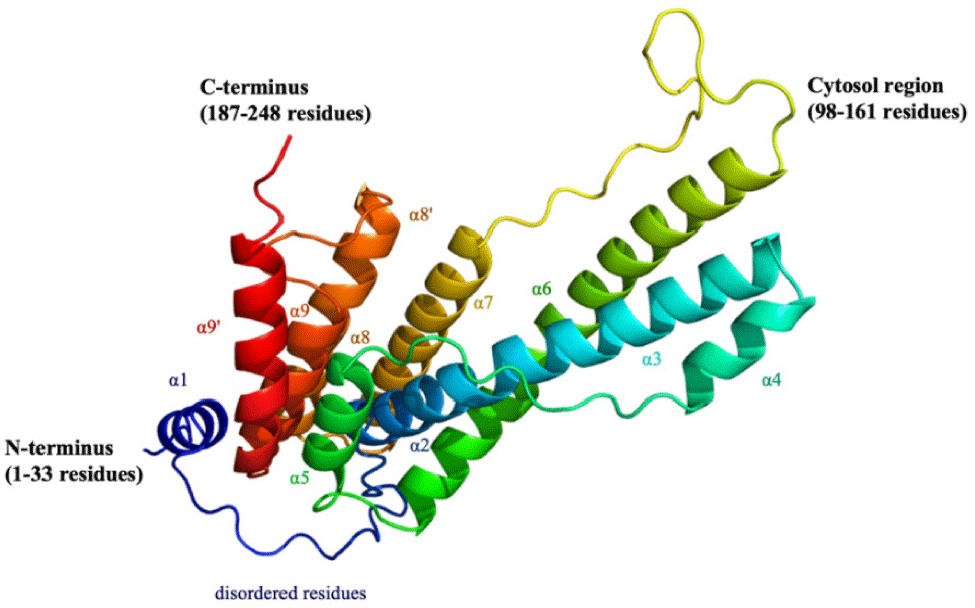
With template-free approach, a total of 150 sequences were obtained from performing multiple sequence alignment. The sequence with the highest identity and coverage (100%) was selected for NS4B protein structure generation. With this sequence, the protein model was generated by using ColabFold 1.5.2 (Fig. 2). In order to ensure the stability and flexibility of the generated protein structure of DENV2 NS4B, MDs was run for this structure for 25 ns.
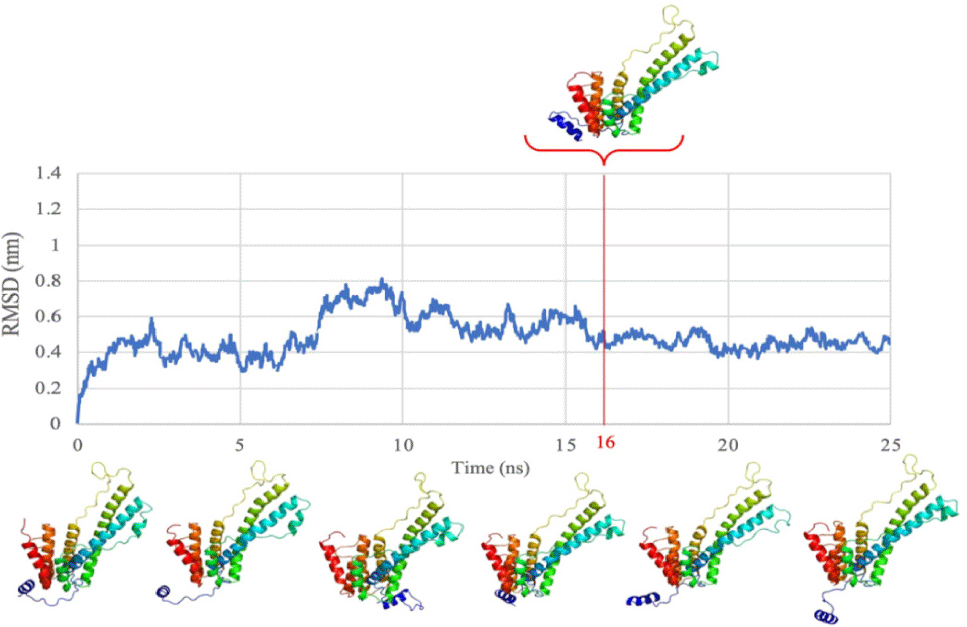
Analysis of the results revealed that the structure reached equilibrium at 16 ns (with the value of RMSD below 2 Å) and maintained stable oscillation until the end of the simulation (Fig. 3). Therefore, the conformation of the NS4B protein of DENV2 at 16 ns was extracted and used for further steps.
Three approaches were used for postdiction of binding site of DENV2 NS4B. First, using CASTp program (based on geometry), 35 binding cavities with their corresponding volumes were identified on the DENV2 NS4B protein. Among them, only cavity A1 was selected since it met the volume requirement over 300 Å3. Secondly, with P2Rank program (based on machine learning), site B1 was chosen as it possessed the highest score of 55.02 and a probability of 0.983. Finally, blind docking identified two binding sites on the NS4B protein: C1 and C2. Among them, cavity C1 was selected because there were 69 out of 73 ligands bound to. Table 2 provided a summary of the important parameters of A1, B1, and C1.
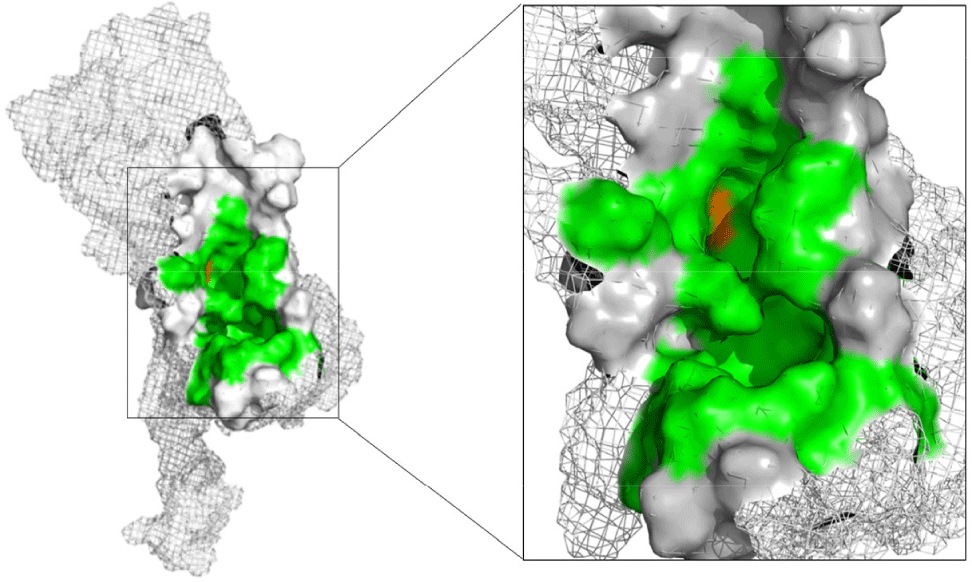
In combination, the results from the three approaches of postdicting binding cavity, the selected binding site consisted of 30 amino acids (Fig. 4). Among them, 29 amino acids (including: Trp38, Tyr41, Ala42, Thr45, Thr46, Thr49, Pro50, Lys86, Asp88, Gly90, Val91, Leu94, Pro162, Glu165, Lys166, Gly169, Gln170, Thr203, Pro209, Gly210, Asn214, Thr215, Thr216, Phe237, Ser238, Lys241, Asn242, Arg247, Arg248) were found in all three methods while 1 amino acid, His117, was retained due to its deep position within the cavity and its involvement in numerous interactions during the blind docking method. Two key residues were postdicted to be Glu165 and Lys166 based on analysis of interactions between protein and 16 reference compounds with their EC50 values ranging from about 10–6 to 8.1 μM in blind docking [4,15,16,38,40,41] (Supplementary Table 3).
Molecular docking was conducted for a total of 286 natural compounds into the DENV2 NS4B protein, resulting in the discovery of 33 compounds with the strong binding affinities (≤–9.0 kcal/mol) from different structural groups: alkaloids (1 substance), flavonoids (15 substances), and terpenoids (17 substances). The alkaloid compound D240 (–9.2 kcal/mol) exhibited strong binding affinity within the binding cavity and created many hydrogen bonds and hydrophobic interactions. However, D240 did not form the same interactions as any reference compounds, and it also did not interact with the key residues Glu165 or Lys166 (Fig. 5). Therefore, D240 was not selected for the next step.
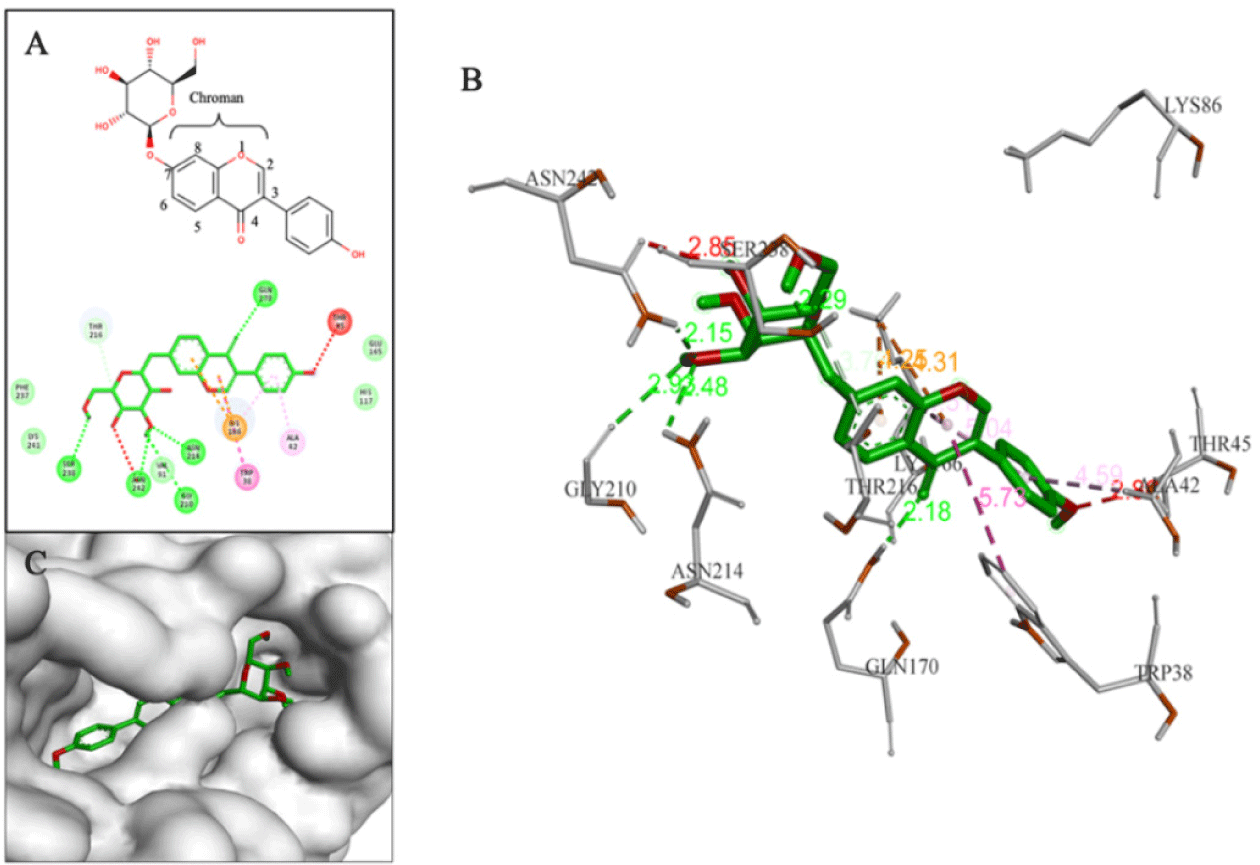
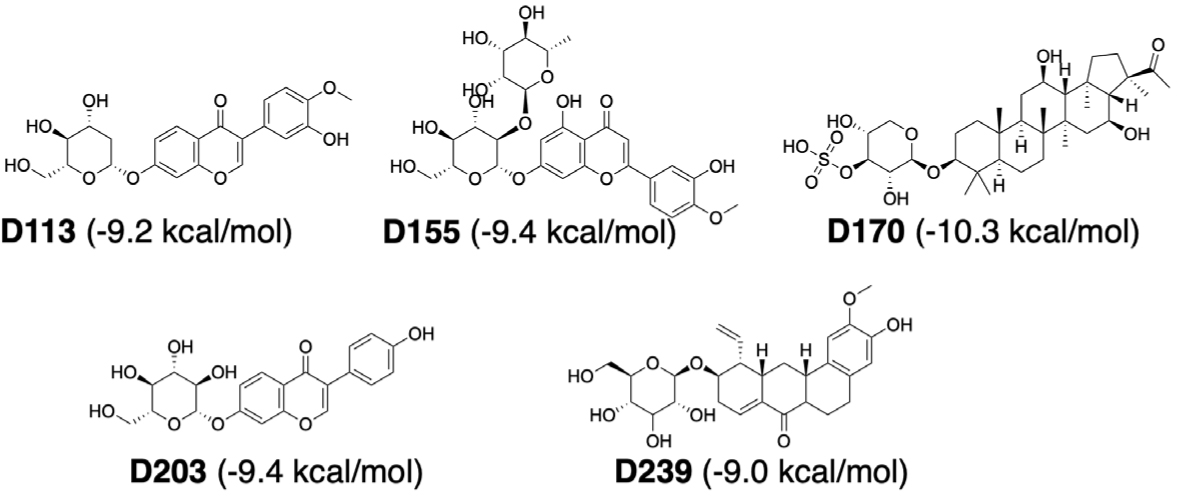
In flavonoid group, all the compounds with good binding affinities were glycosides, including 3 structural groups: eu-flavonoids (9 substances), iso-flavonoids (5 substances) and neo-flavonoids (1 substance). Although these phytochemicals were capable of forming numerous hydrogen bonds and hydrophobic interactions with residues in binding sites, only D113, D155, and D203 had interactions with key residues or similar to interactions observed with the reference compounds. D113 (–9.2 kcal/mol) was classified as an iso-flavone compound belonging to the iso-flavonoid group with a phenyl branch at position C3 and a glycol group at position C7. This compound formed a hydrogen bond between the hydroxyl group (-OH) on the glycol part and hydrogen in Glu165. It also had many hydrophobic interactions with amino acids, including Lys166 (Fig. 6).
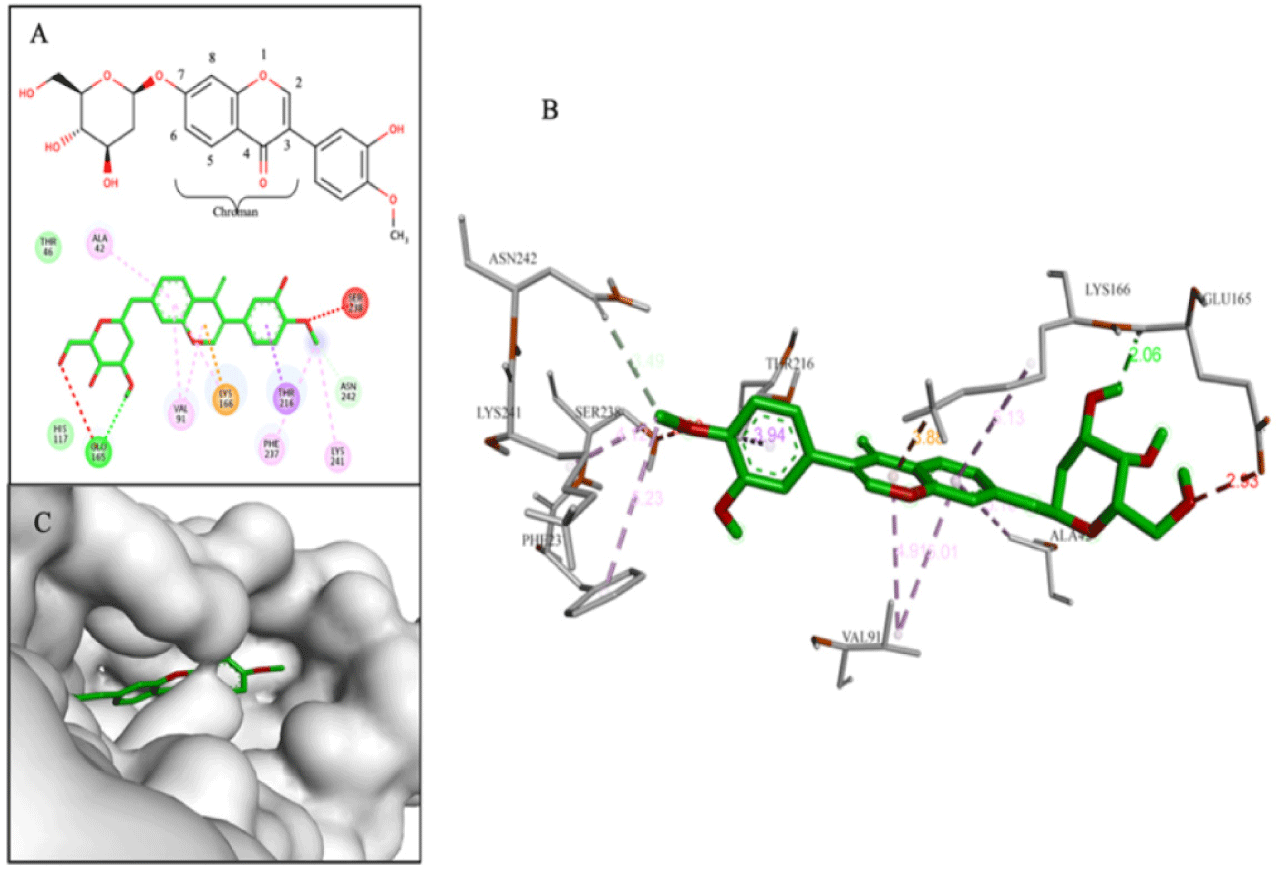
Compound D155 (–9.4 kcal/mol) belonged to the eu-flavonoid group and was classified as a flavone compound. It had a phenyl branch at C2 and two glycol substituents at position C7 of chroman skeleton. This compound interacted with the binding cavity through hydrogen bond with Glu165 and π-Alkyl bond between the chroman ring and Lys166 (Fig. 7).
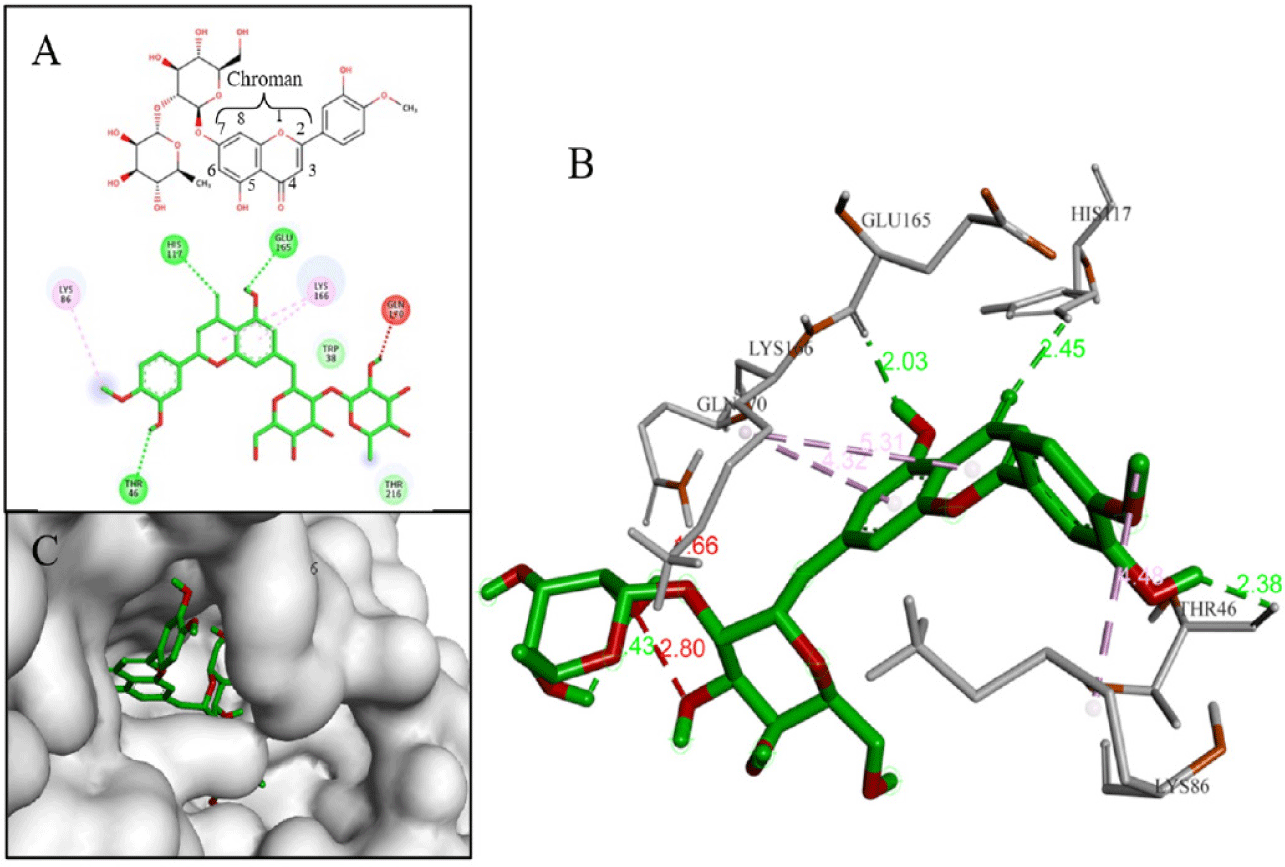
Compound D203 (–9.4 kcal/mol) was an iso-flavone compound. D203 created multiple hydrogen bonds with amino acids (Gln170, Gly210, Asn214, Asn242) at the hydroxyl groups (-OH) on the phenyl ring and the glycol part. These interactions were similar to those observed for the reference compound F35 (NITD-688), which was a pan-serotype inhibitor and exhibited anti-DENV2 activity with an EC50 of 0.008 µM [4]. Additionally, hydrophobic interactions were also analyzed between D203 and three amino acids Trp38, Ala42, and Lys166 (Fig. 8).
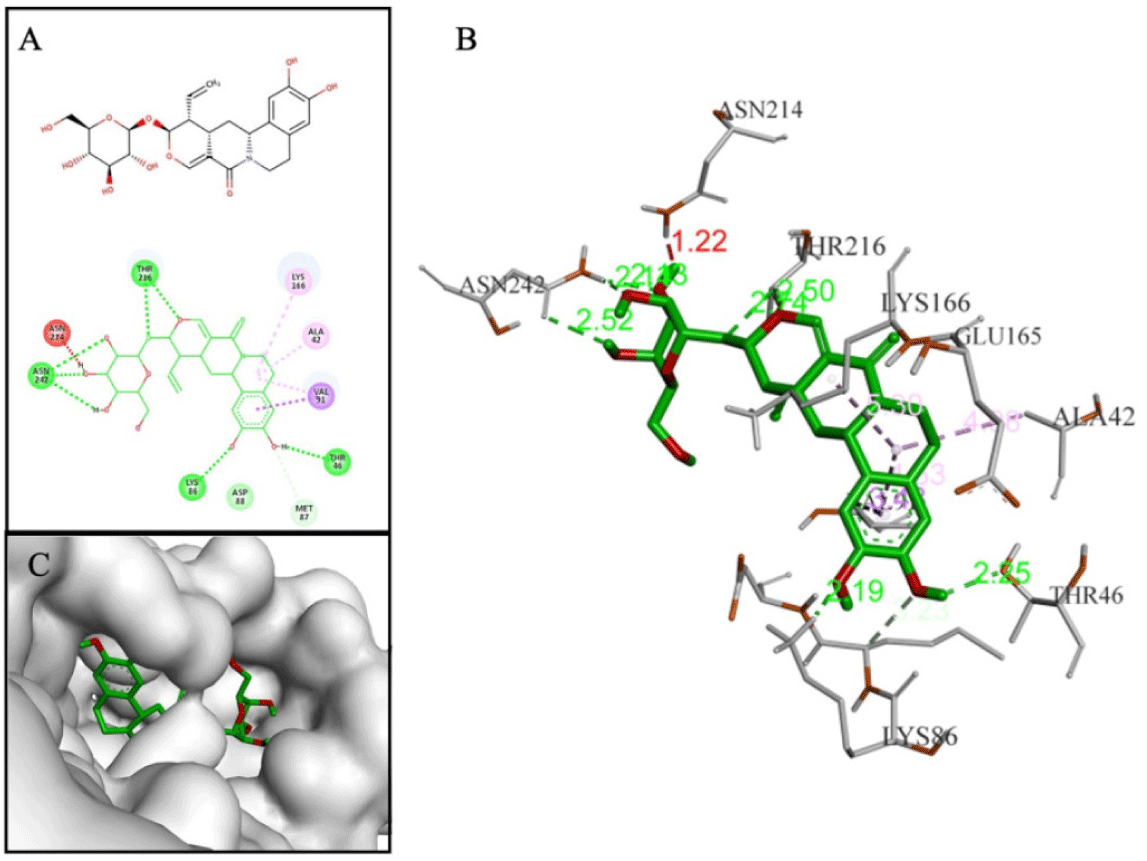
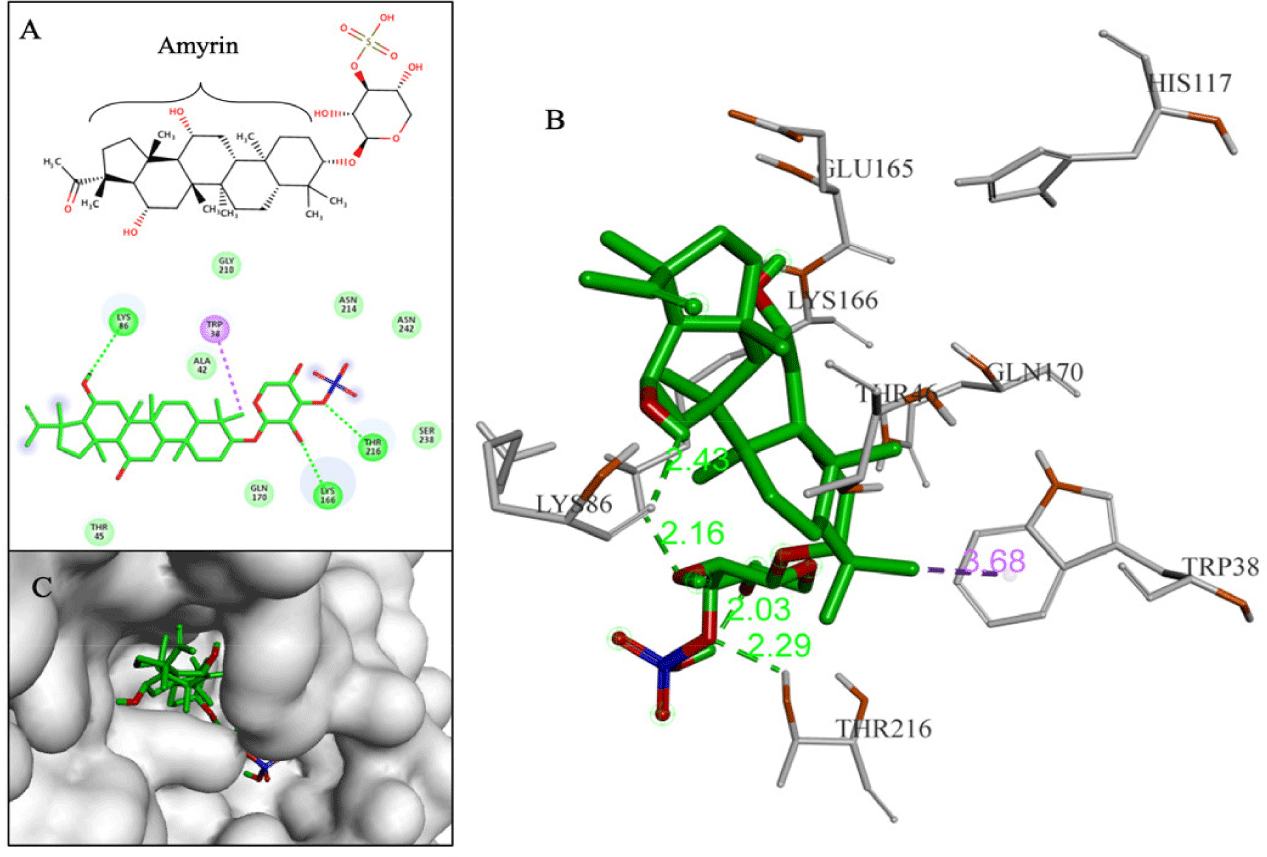
Terpenoids with good binding affinities belonged to 3 groups: sesterterpenes (3 substances), triterpenes (12 substances) and saponins (2 substances). Among them, D170 and D239 (2 saponins) were the two substances chosen as the most promising. Compound D170 (–10.3 kcal/mol) was a saponin that consisted of a short glycol part connected to a sulfate group. This arrangement led to the formation of a hydrogen bond between the ether group (-O-) and hydrogen in the hydroxyl group (-OH) on Thr216. The aglycon part of D170 included an amyrin skeleton with multiple methoxy substituents, resulting in hydrophobic interactions with the amino acid Trp38. Notably, these interactions closely resembled those observed with the F42 reference compound (AM404), which was an active metabolite of paracetamol and exhibited anti-DENV2 activity with an EC50 of 3.6 µM [4]. Additionally, D170 also interacted with Lys86 and Lys166 through hydrogen bonds (Fig. 9).
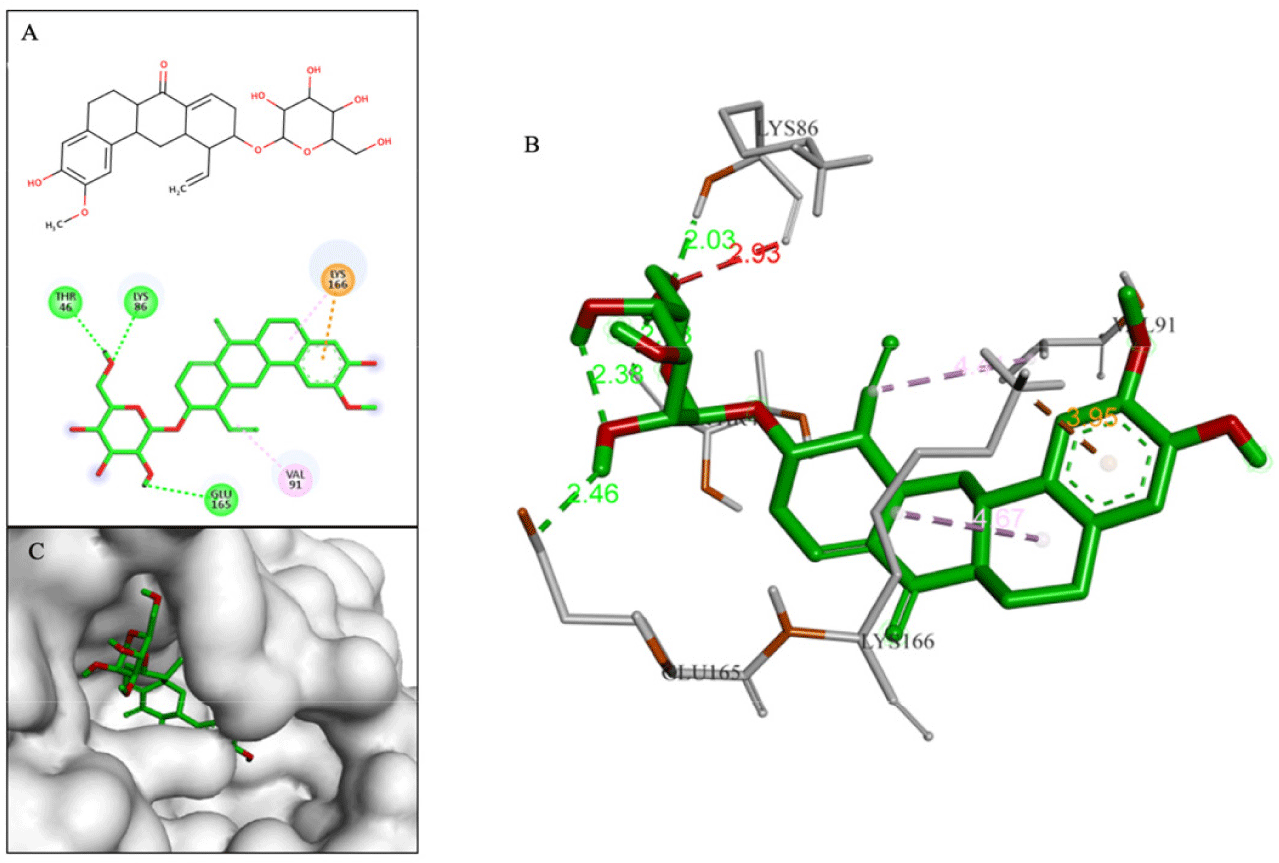
Compound D239 (–9.0 kcal/mol) with saponin structure demonstrated significant interactions with amino acids in the binding site. The glycol part of D239 formed multiple hydrogen bonds, particularly with key residue Glu165. Additionally, the aglycon part of D239 contained an aromatic ring that was deeply inserted into the cavity, resulting in a hydrophobic interaction with key residue Lys166, as depicted in Fig. 10.
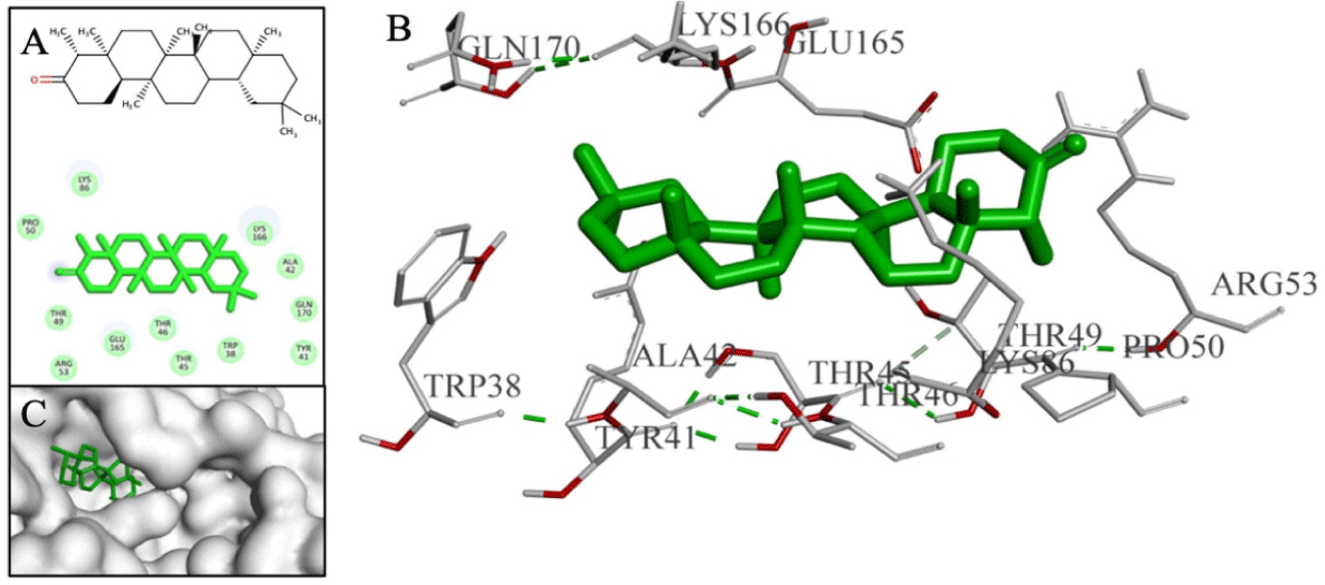
Additionally, the sesterterpene and triterpene compounds, typically D126 with binding affinity of –11 kcal/mol, exhibited strong binding affinities but were unable to form interactions with the residues within the binding cavity (as shown in Fig. 11). As a result, they were excluded from the final screening results.
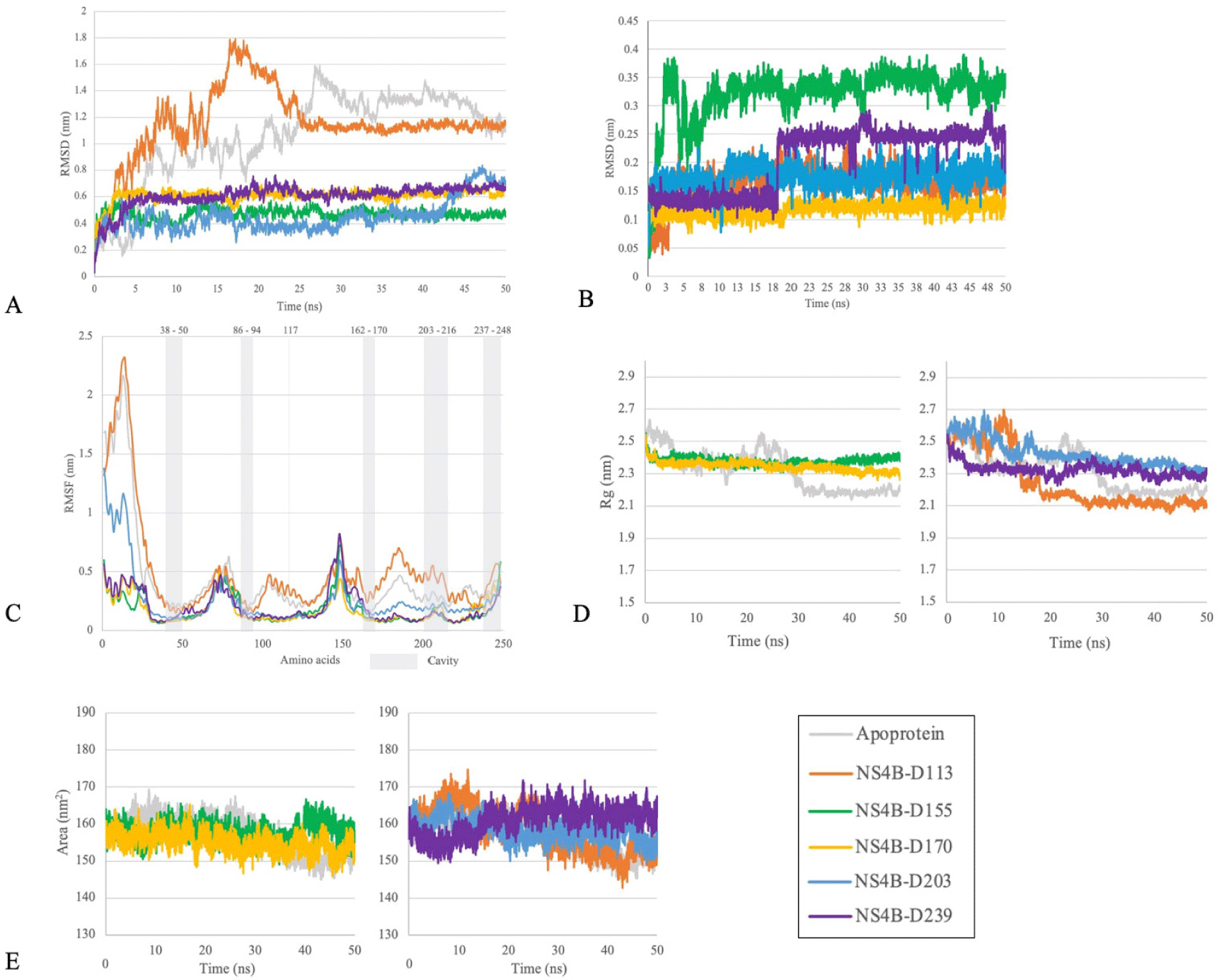
The results of the simulation process of 5 complexes between protein-ligand (D113, D155, D170, D203, D239) and apo-protein were postsented in Fig. 12. By examining the RMSD, RMSF, Rg, and SASA values of these complexes compared to apo-protein and the stability of each ligand through their heavy atoms, our study highlighted the ability to form stable complexes of two compounds D155 and D170 during 50 ns of simulation. These two protein-ligand complexes exhibited both protein and ligand RMSD deviations of less than 0.2 nm after 25 ns and the oscillation remained stable until the end of the simulation. Additionally, the RMSF values of the residues within the binding cavity were lower than the values observed for the apo-protein. Moreover, both Rg and SASA values were more stable compared to the apo protein. Therefore, the two complexes of the NS4B protein with the ligands D155 and D170 were selected for further hydrogen bond occupancy analysis (Table 3) and long-time scale MDs (Fig. 13).
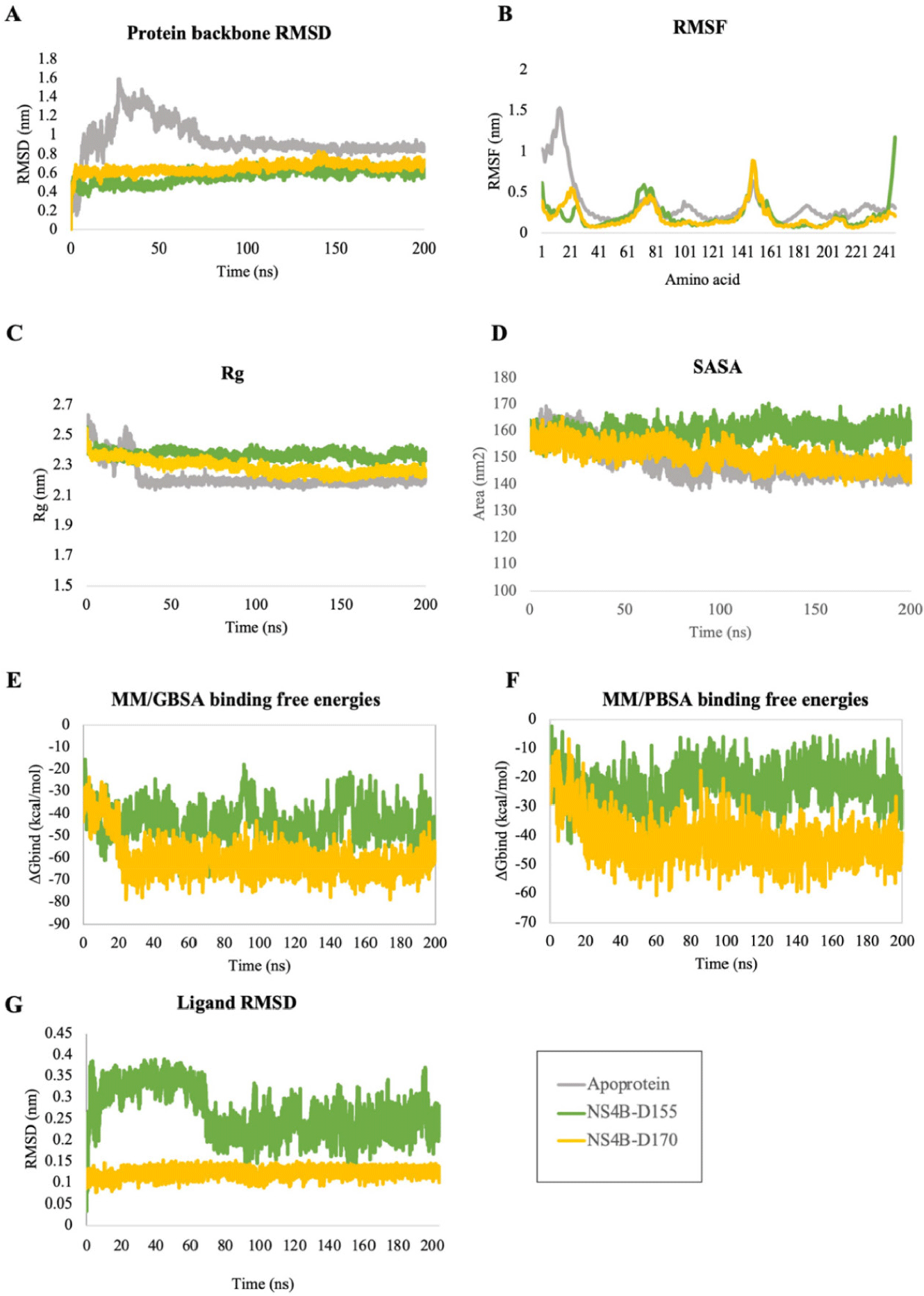
A rapid assessment of the drug-like and ADMET properties of these 5 ligands was carried out by using the ADMETlab 3.0 web server [54] and the postdicted results were postsented in the Supplementary Table 5. The results showed that all compounds satisfied at least one drug-likeness rule, with no pan assay interference compounds (PAINS) alerts and no serious toxicity. However, D170 performed good absorption compared to other compounds.
The binding free energy analysis for the NS4B protein and its complexes with the top hit ligands, D155 and D170, were derived from 200 ns MDs trajectories (from frame 1 to 20,001 with interval of 10). The ΔGblind values and energy components were postsented in Table 4. Their fluctuations over time were depicted in Fig. 13. In the first 25 ns of the simulations, the binding free energies tended to decrease because the complexes progressed toward equilibrium. After reaching equilibrium, the free energy remained negative until the end of 200 ns of simulation. These results indicated that the interactions between the two ligands and the NS4B protein were formed and stayed stable throughout the simulation.
4. DISCUSSION
Phytochemicals have long been used in traditional medicine to treat various diseases and have been shown to inhibit viral replication and transcription [55]. Various plant-derived products have been extensively studied against viruses such as herpes, human immunodeficiency virus, influenza, hepatitis, and SARS-CoV-2 [55]. However, there is still limited exploration of phytochemicals for the inhibition of viruses like dengue virus [55]. Therefore, in this study, structure-based virtual screening through molecular docking and MDs was conducted to evaluate 286 natural compounds targeting the NS4B protein of DENV2 to identify potential anti-dengue agents.
Despite the lack of crystal structure of NS4B protein of DENV2, its topology has been investigated so far. In postvious NMR study, an 11-helix model was proposed for the secondary structure of the DENV NS4B protein with 6 of these helices remaining membrane-buried [56]. The N-terminal region comprised a small helix (α1) and disordered residues [56]. The C-terminal region contained four small helices (α8, α8’, α9, α9’), where α8 and α8’ were not fully membrane-buried, α9 and α9’ could interchange for mobility [56]. Similar to these, the ColabFold model postdicted a total of 11 helices, out of which one small helix in the N-terminal and four helices in the C-terminal region (Fig. 2). In postvious studies on the unstructured biology of proteins of dengue virus, the NS4B protein of DENV2 exhibited a disorder propensity of 14.5% [57]. These included disordered N- and C-terminal tails and the cytosolic loop region [57], which were considered as intrinsically disordered protein regions (IDPRs) of NS4B protein of DENV2. Long MDs exceeding 1 µs for the C-terminal in an aqueous environment revealed similar findings [58]. IDPRs exist as dynamic conformational ensembles with varying levels of residual structure over simulation time, which affected the RMSD fluctuations, ranging from collapsed (molten globule-like) to partially collapsed (post-molten globule-like), and even highly extended (coil-like) conformations [57]. These regions were postdicted to be unsuitable for stable drug binding. Therefore, to reduce complexity and computational resources, the structure at the single frame of NS4B protein, after both the non-IDPRs and IDPRs of the protein had reached a stable state, was extracted for further analyses. In this study, the postdicted NS4B protein was run MDs to obtain the stable conformation and more accurate structure of NS4B. This structure reached equilibrium stage from 16 ns of MD, thus the NS4B conformation at the 16 ns frame was the stable conformation. Clustering analysis also showed that the conformation at the 16 ns also belonged to the first repostsentative structure cluster of NS4B. Thus, this structure of NS4B at 16 ns was used for virtual screenings in the next stage.
The most stable MD conformation and the best binding pocket are two different aspects. The most stable MD conformation does not necessarily correspond to the best binding pocket. Therefore, utilizing the stable conformation of the NS4B protein structure, the best binding pocket was identified by using a combination of three methods based on distinct principles: (1) CATSp, which identified pockets based on the 3D structure of the protein and spatial geometry, (2) traditional machine learning that utilized the random forest algorithm implemented in P2Rank, and (3) blind docking of known bioactive compounds across the entire target protein. The optimal binding pocket was selected by integrating the results from these three approaches.
73 compounds possessing the activity against DENV2 or the NS4B protein of DENV2 from 30 articles [4,15–43] (Structure and corresponding EC₅₀ values (μM) listed in Supplementary Table 1 were selected for docking. Among the reference compounds, the compound F11 corresponds to JNJ–A07 (as shown in Supplementary Table 1). It is a pan-serotype dengue virus inhibitor targeting the NS3–NS4B interaction [1]. In the original study conducted by Kaptein et al. in 2021 [1], JNJ–A07 was evaluated for its antiviral activity against DENV2 in six cell lines and various genotypes of all four DENV serotypes. This compound exhibited antiviral activity at nano- to picomolar concentrations across various cell lines and against the diversity of known genotypes and serotypes of the dengue virus, including DENV2. Although the EC50 value of F11 for DENV2 was not postcisely determined, the activity of JNJ–A07 was also confirmed in a mice model of DENV2 infection [1]. For this reason, F11 was used as a reference compound for this study.
Through the criteria including ligand binding affinities, binding modes and binding interactions between the NS4B protein and 33 compounds, the top five promising compounds were identified as D113 (–9.2 kcal/mol), D155 (–9.4 kcal/mol), D170 (–10.3 kcal/mol), D203 (–9.4 kcal/mol), and D239 (–9.0 kcal/mol) (Fig. 14). These compounds shared two key characteristics, including the postsence of glycol group with multiple hydroxyl (-OH) substituents and a bulky aglycon group that increased the overall hydrophobicity of the molecules. The glycol substituents enabled the formation of hydrogen bonds between the compounds and the surrounding hydrophilic residues within the binding cavity of the NS4B protein. Additionally, the hydrophobic aglycon group allowed the compounds to attach deeply and interact with the residues in the binding pocket, especially the key residue Lys166, which likely contributed to their strong binding affinities.
The molecular docking analysis showed that compound D155 created stable interactions including hydrogen bonds with His117 and Glu165, as well as hydrophobic interaction with Lys166. After simulation, these hydrogen bond interactions remained consistently stable with high occupancy, reaching 188.15% for the interaction with His117 and 267.41% for the interaction with Glu165. However, the hydrophobic interaction with Lys166 was replaced by hydrogen bond at a frequency of 148.42%. Furthermore, after MDs, this compound also formed new additional hydrogen bonds with other amino acids, such as Lys24, Trp38, Thr45, Lys86, and Thr216, with a frequency greater than 70%. On the other hand, D170 demonstrated hydrogen bonds with Lys86, Lys166, and Thr216 during molecular docking. However, after the MDs, only the hydrogen bond with Thr216 was maintained and new hydrogen bond was formed with Glu165 with occupancy of 198.98% and 225.09%, respectively. In particular, binding free energy calculation revealed that both complexes had negative binding free energies, regardless of the two calculation approaches, indicating their ability to form stable complexes with the NS4B protein until the end of the simulations. Combining the results from docking, MDs, and ΔGblind values, D155 and D170 were selected as the most potential NS4B inhibitors.
Currently, there has been no investigation on the potential of compounds D155 and D170 to inhibit the NS4B protein or DENV strains. However, in postvious studies, these compounds have shown multiple promising activities. D155 (Neodiosmin) is a type of flavonoid that can be derived from the butanol extract of the Valeriana hardwickii plant, which belongs to the Valerian family (Valerianaceae) or citrus plants (Citrus bergamia, Citrus aurantium, Cirus australasica…). It has shown promise to be an anti-SARS-CoV-2 agent (in silico and in vitro) [59] and antioxidant (in vitro) [60]. D170 (Spergulin A) is a saponin extracted from the ethanol extract of Glinus oppositifolius Molluginaceae. It has shown the ability to resist the parasite Leishmania donovani (in vitro) [61] and antibacterial (Escherichia coli, Haemophilus influenzae, Staphylococcus aureus, etc.) (in vitro) [62].
In addition to the promising findings regarding the inhibitory potential of two natural compounds against the NS4B protein of DENV2, the study has some limitations that should be noted. In this research, the structural analysis of the NS4B protein of DENV2 was obtained by postdicted and then used for MDs to obtain more accurate and equilibrium and stable conformational structure, However, MDs were investigated in an aqueous environment, which does not account for the influence of the lipid bilayer in maintaining the equilibrium of the protein, particularly in IDPRs. Thus, the natural compound database used for screening in this study included only an in-house collection of over 200 natural compounds. Expanding this library to include a broader range of natural compounds could enhance the scope of future investigations targeting the NS4B protein of DENV2. Finally, the inhibitory potential of the compounds was postdicted only for the monomeric state of the NS4B protein, without considering the effects of interactions between NS4B and other proteins, including NS3, NS4A, NS5, NS1, and the dimeric state of NS4B.
5. CONCLUSION
In this study, molecular docking and MDs approaches were applied to discover potential inhibitors of NS4B protein of DENV2. Due to the lack of crystal structure, template-based and template-free approach were applied to postdict the tertiary structure of DENV2 NS4B protein. The obtained 3D structure of NS4B reached equilibrium after 16 ns of MDs. Based on three binding site postdiction methods, a ligand binding cavity of NS4B consisting of 30 amino acids was identified. Virtual screening of natural compounds by molecular docking and analyzing stability by MDs and ΔGblind values showed that two compounds D155 (Neodiosmin) and D170 (Spergulin A) had potential in inhibiting NS4B protein of DENV2. Overall, this study suggests that Neodiosmin and Spergulin A can be further investigated to be used as potential therapeutic treatment on DENV2 NS4B protein.