






1. INTRODUCTION
Skin aging is a complicated phenomenon which is a combination of natural aging and photo-aging [1]. It is related to progressive loss of skin structural integrity and physiological function leading to visible wrinkles and skin sagging on the skin’s surface affecting aesthetics [1]. This process could be due to the change of various internal factors, for example hormone, cellular metabolism, genetics and/ or external factors such as the exposure of sunlight (i.e., photo-aging), pollution, and chemical toxins [1]. Of which, UV radiation exposure could cause the production of reactive oxygen species (ROS) which is a dangerous agent to induce the skin aging prematurely by changing of production of collagen, elastin, melanine, etc through the inhibition and/ or activation of some enzymes such as hyaluronidase, elastase, matrix metalloproteinase in the extracellular domain of the skin [1].
Matrix metalloproteinases (MMP) are a endopeptidase family or matrixins that are zinc (II)-dependent proteases [2]. There are 23 MMPs (MMP-1 to MMP-23) belonging to five main groups including collagenase, gelatinase, stromelysins, matrilysins, membrane MMPs (MT-MMP), and other MMPs [2]. Among these enzymes, MMP-1 (collagenase), MMP-3 (stromelysin-1) and MMP-9 (gelatinase) are three major enzymes responsible for degradation of various extracellular protein components of skin such as degrading collagen in the epidermis of skin surface [2]. Thus, MMP-1, MMP-3 and MMP-9 are considered potential targets for anti-aging drug discovery.
MMP family have some common structures [2]. Typically, the structures of MMPs include a propeptide region (77-87 amino acids), a catalytic metalloproteinase domain (about 170 amino acids), a linker peptide (hinge region) and a hemopexin domain (200 amino acids) [2]. The catalytic zone contains two Zn2+ ions for structural and catalytic and three calcium ions (Ca2+); in which the zinc ion for catalytic binding with three histidine and 3 Ca2+ ions [2]. In the catalytic zone there are active zones, which interact with substrates and inhibitors. These parts include S1, S2, S3,… Sn on the left side of Zn2+ ion and S1’, S2’, S3’,… Sn’ on the right of Zn2+ ion [2]. In particular, the pockets on the right are considered to be the places of binding inhibitors as they provide better inhibitory effects and the pockets on the left are less interested because of a relatively modest inhibitory capacity. The S1’ pocket was known to be important in determining the MMPs selectivity due to the difference in size, hydrophobicity and flexibility among MMPs [2].
For the MMP-1, this S1’ pocket is quite shallow and less hydrophobic. However, the amino acid Arg214 at the bottom of this pocket is flexible, which can be displaced during ligand binding to create a larger bound pocket. For the MMP-9, the S1’ pocket is wider and more hydrophobic, with the bottom of this pocket being the inflexible amino acid Arg424, making it difficult for the ligand to attach with long branches. The MMP-3 has the widest S1 ‘pocket, as an open channel, suitable for bulky and highly hydrophobic ligands [2].
Until now, there are some researches about natural compounds such as alkaloids, flavonoids, phenolic compounds for anti-aging activity, especially for MMPs inhibition. For example, aloin A, B of Aloe vera [3]; xanthorrhizol in the extract of Curcuma xanthorrhiza [4], diosgenin of Dioscorea villosa [5], oil extract from Camellia japonica [6] and fruit extract from Emblica officinalis [7] inhibited MMPs enzymes. Morus alba Moraceae has been used as traditional medicine with some bioactive compounds for anti-inflammatory, antioxidant and neuroprotective activities [8]. The in vitro effects of Morus alba extract were also reported on the MMP-1, MMP-9 [9] and MMP-13 [10] for the treatment of osteoarthritis and periodontitis. Unfortunately, there is no information about biological activity of Morus alba phytochemicals as potential anti-aging agent. Therefore, the present study investigated the anti-aging potency of natural compounds in the Morus alba extract through their binding interactions with the MMP-1, MMP-3 and MMP-9 enzymes related to skin aging process by a combination of molecular docking and molecular dynamics simulations approaches.
2. MATERIALS AND METHOD
Molecular docking between three receptor targets, namely MMP-1 (PDB id: 966C [11]), MMP-3 (PDB: 1G4K [12]) và MMP-9 (PDB: 2OW0 [13]) and 9 phytochemical compounds found in the Morus alba extract was conducted in AutoDock 4.0 [14] to explore the binding affinity, binding pose as well as binding interactions of the targets with respective ligands. Briefly, three targets were retrieved from the protein data bank (https://www.rcsb.org). The crystal structures of MMP-1 (PDB: 966C), MMP-3 (PDB: 1G4K) and MMP-9 (PDB: 2OW0) were downloaded in format .PDB file (Table 1), and then removed waters and retracted the co-crystallized ligands from the experimental structures and kept the ion Zn2+ in the active sites of the protein targets. The targets were minimized by using CHARMM 36 force field in Discovery Studio 2016 [15], and save the monomers in .PDB file. These protein targets were subsequently added polar hydrogens, Gasteiger charges by using Autodock Tool 1.5.6 [14] and save in the .PDBqt file.
| Protein | PDB id | Resolution | Structure type | Selected chain for docking | Co-crystallized ligand | Reference |
|---|---|---|---|---|---|---|
| MMP-1 | 966C | 1.9 Å | Monomer A | Monomer A | N-hydroxy-2-[4-(4-phenoxy-benzenesulfonyl)-tetrahydro-pyran-4-yl]-acetamide | [11] |
| MMP-3 | 1G4K | 2.0 Å | Trimer A, B, C | Chain B | 5-methyl-5-(4-phenoxy-phenyl)-pyrimidine-2,4,6-trione | [12] |
| MMP-9 | 2OW0 | 2,.0 Å | Dimer A, B | Chain A | N-[(4’-iodobiphenyl-4-yl)sulfonyl]-d-tryptophan | [13] |
The optimization parameters for docking were updated in files protein. PDBqt and AD4_parameters.dat including for Zn2+: Rii = 0,87 Å (Rii was the sum of van der Waals radius of two similar atoms), epsii ε = 0,35 kcal.mol-1 (ε was the depth of van der Waals) and volume of atomic solvation vol [16].
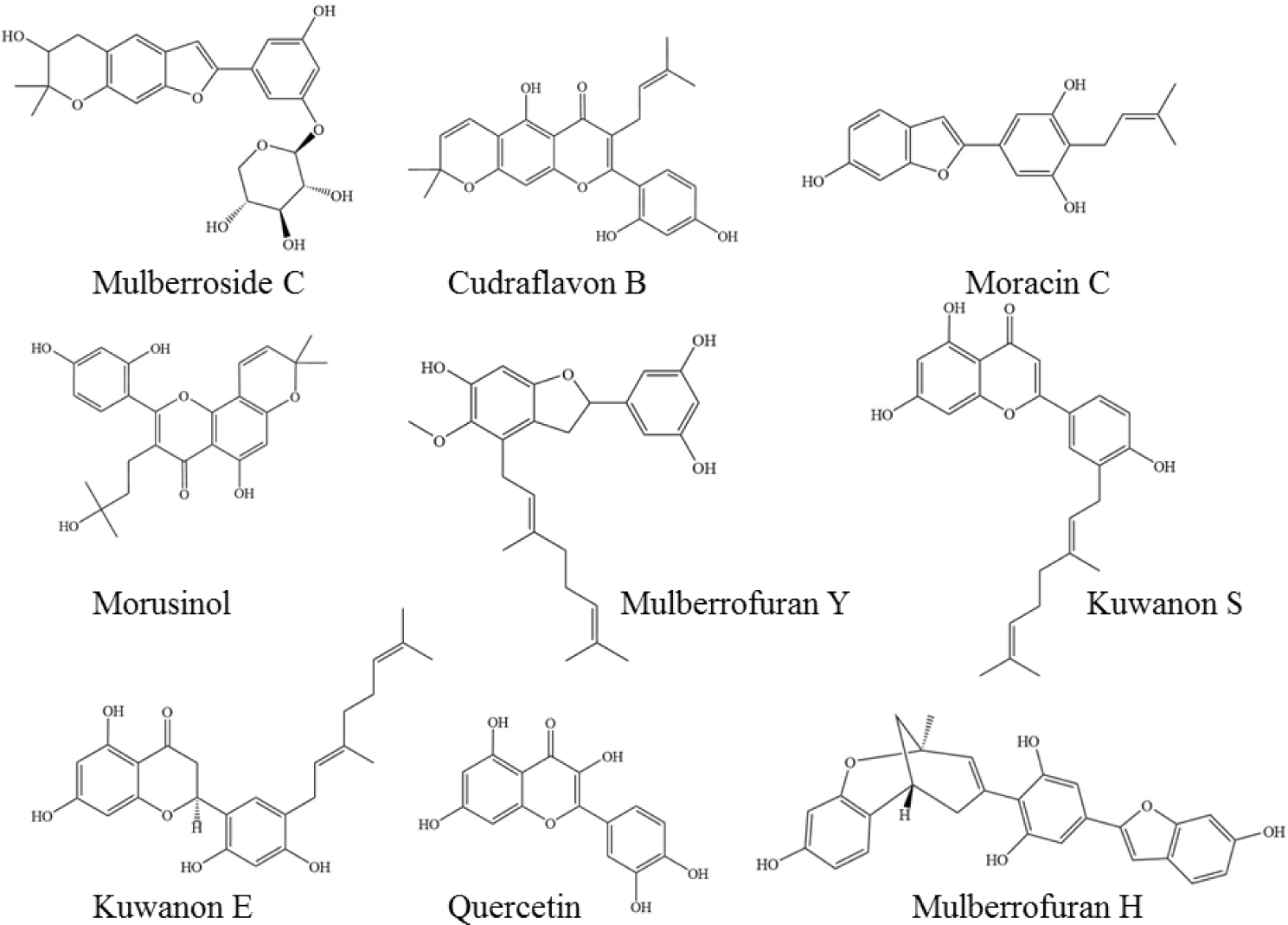
The nine phytochemicals in the Morus alba extract included 5 prenyl flavonoids (Kuwanon E, Kuwanon S, Quercetin, Morusinol and Cudraflavon B), 2 benzofurans (Mulberrofuran Y and Mulberrofuran H), Moracin C and Mulberroside C. These compounds were selected for the study based on the similar structures to the co-crystallised ligands in the MMP enzymes. The 2D structures of compounds (Figure 1) were downloaded from PubChem database (https://pubchem.ncbi.nlm.nih.gov/) in the format .sdf file and minimized energy by using ChemDraw 3D 16.0 and save in the format .PDB. Following, the targets and ligands were prepared using Autodock Tools 1.5.6 under the setting parameters of grid boxes (Table 2) and saved in .gpf. Autogrid 4.0 converted .gpf to .glg file. Finally, molecular docking was carried out using AutoDock 4.0 with the Lamarckian genetic algorithm and input files .glg and .dpf. The docking results (in files .dlg) were analysed using Discovery Studio 2016 và MOE 2015.10 including binding affinities, different conformational ligands and the binding interactions of ligands and protein targets.
Molecular dynamics simulations (MDs) were used to investigate stability and flexibility of protein-ligand complex under the influence of temperature, pressure, and solvent. MDs were conducted for protein without ligand (apo protein) and protein-ligand complex for 20 ns by using GROMACS 2019 software. From the docking results, choosing the best conformation of the best binding compound and then adding hydrogens to it by Avogadro ver 1.2.0 software were performed. The topology of the ligand was generated by CGENFF with CHARMM 36 force field. The complex was placed in a simulation box (12 surface polyhedrons) and contained the water solvent (TIP3P model) and a distance of 1.0 nm from the box edge. The sodium cation and chloride ion were added to neutralise the system. The complex was energy minimized to eliminate the negative interactions and the system was balanced under the “isothermal-isobaric” NVT and NPT conditions (N: number of particles, V: volume, T: temperature 300 °K, and P: pressure 1 bar). Berendsen thermostat and Parrinello-Rahman barostat were used for maintaining the temperature and pressure. The equilibration was run during 1000 ps for each NVT and NPT systems. The production was for 20 ns and the result was taken every 0.01 ns and evaluated based on the values of RMSD (Root-mean-square deviation), RMSF (Root-mean-square fluctuation), Rg (Radius of gyration), percentage of hydrogen bond occupancy and hydrophobic interactions and SASA (Solvent-accessible surface area). The parameters were calculated by GROMACS, and the results were analysed using VMD version 1.9.3.
3. RESULTS
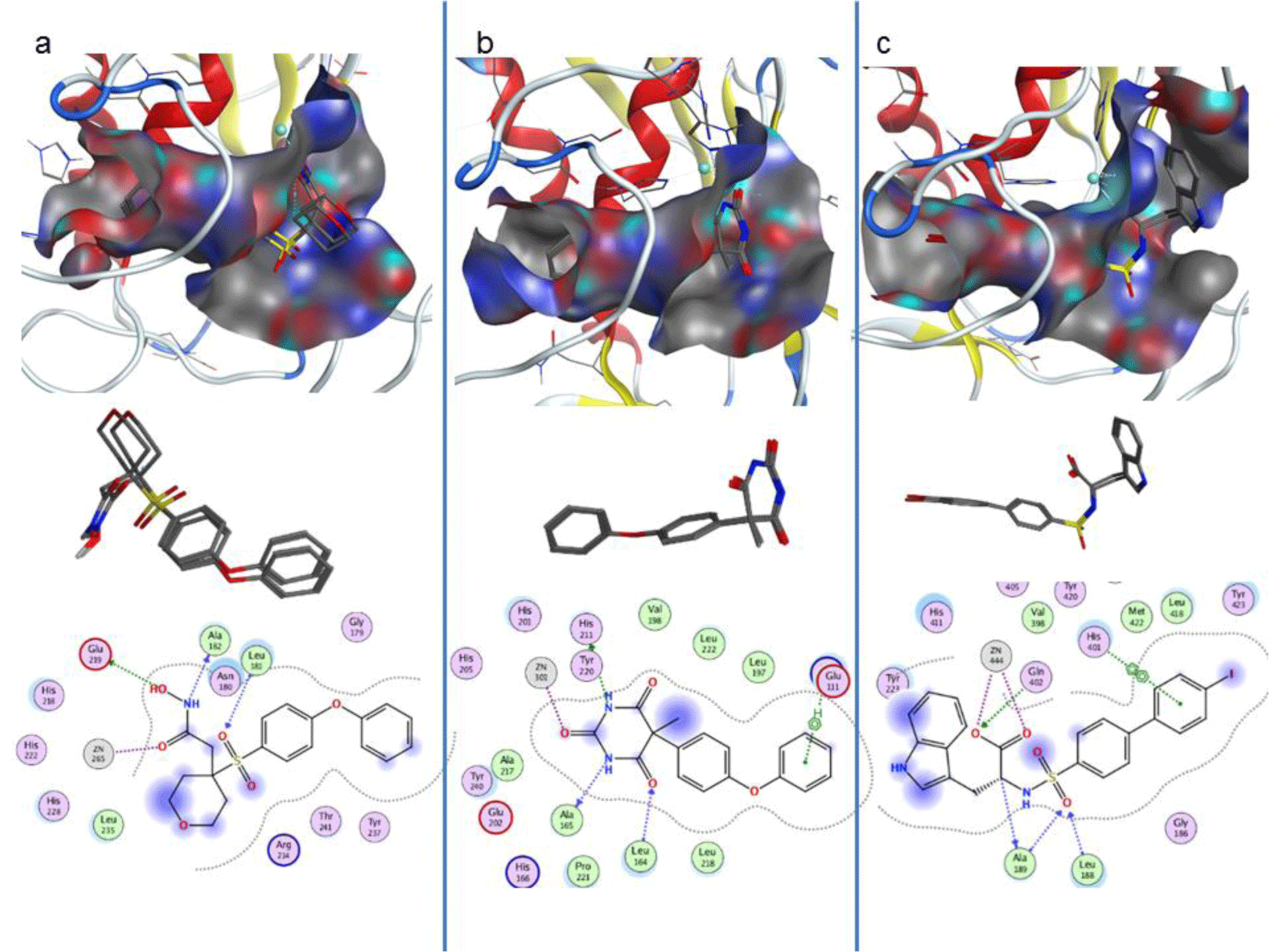
Molecular docking was carried out for the co-crystallised ligands in the three experimental structures to evaluate the reproducibility of Autodock 4. The results showed that these native ligands obtained good binding affinities of -7.71; -8.19 and -8.98 kcal.mol-1 into the three targets, MMP-1, -3 and -9, respectively. The root-mean-square-deviations of conformations of ligand after docking and the native ones were about 0.2 to 0.6 Å. The active sites of the targets were confirmed including the co-crystallised ligands, ion Zn2+ at the center with 3 histidines along with the key residues Leu181, Ala182 và Glu219 of MMP-1; Leu164, Ala165, Ala167 and Glu202 of MMP-3; and Leu188, Ala189, Gln402 of MMP-9, respectively (Figure 2). The binding interactions of the experimental structures were also reproduced after docking (Table 3).
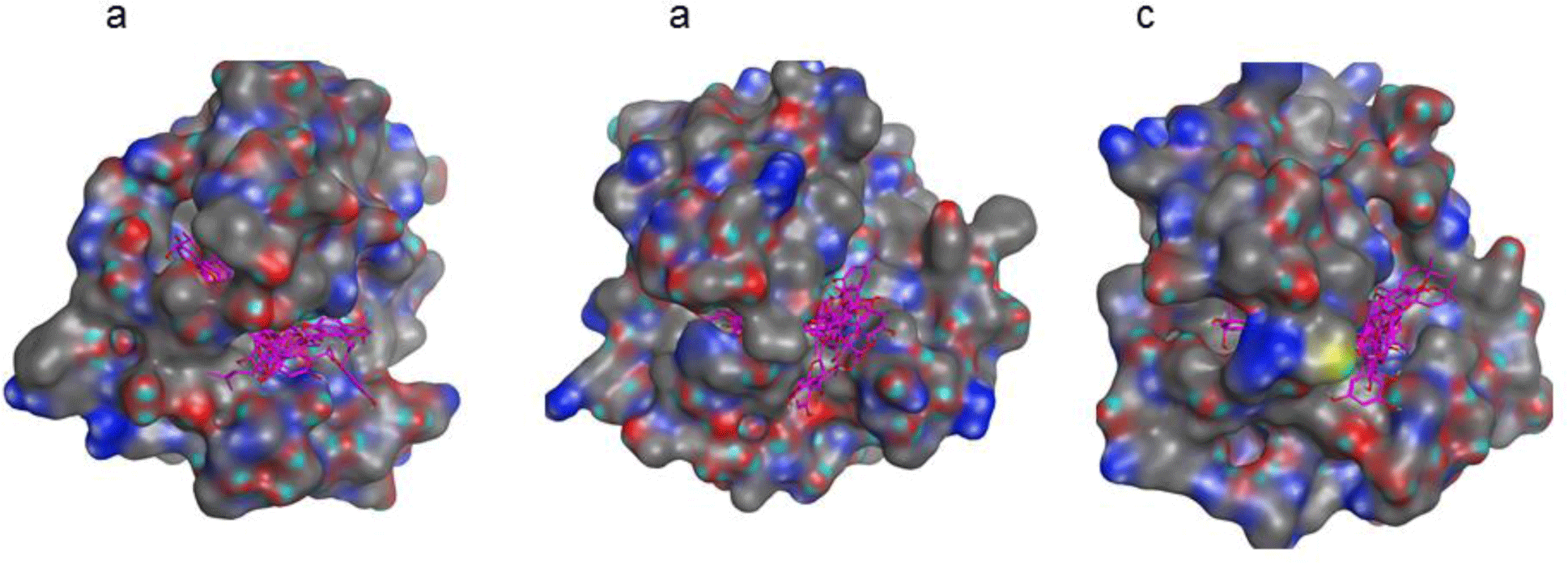
Molecular docking of 9 compounds, kuwanon E, kuwanon S, quercetin, morusinol, cudraflavon B, mulberrofuran Y, mulberrofuran H, moracin C and mulberroside C into three targets, MMP-1 (PDB: 966C), MMP-3 (PDB: 1G4K) and MMP-9 (PDB: 2OW0) was carried out using Autodock 4.0. The docking results demonstrated that all nine compounds were fitted well into the active sites of all proteins with the good binding affinities (Figure 3 and Table 4). Mulberrofuran H, moracin C and kuwanon S docked well on all MMP-1, MMP-3, MMP-9 proteins with the binding affinities lower than -7.0 kcal.mol-1. Among the investigated compounds, mulberrofuran H was selected as the hit compound with the best binding affinities on three protein targets: -8.23 kcal.mol-1 for MMP-1; -8.87 kcal.mol-1 for MMP-3 and -8.34 kcal.mol-1 for MMP-9. Some top compounds with their interactions with three proteins were listed in Table 5.
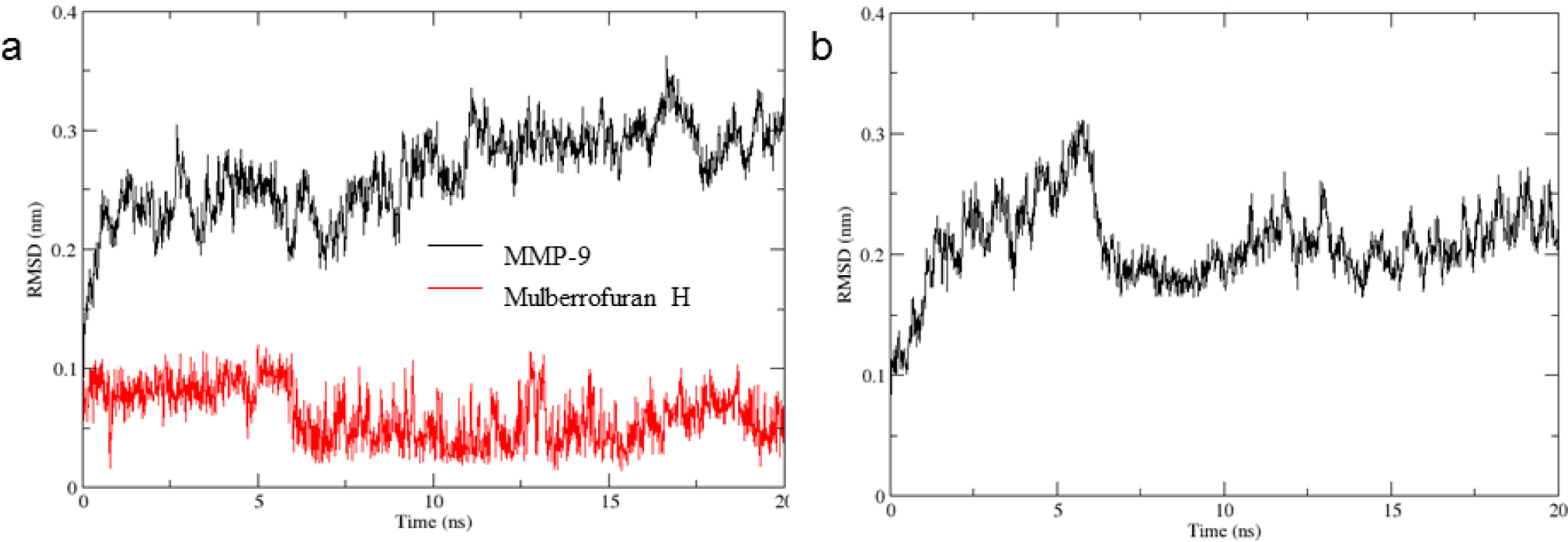
The best complex of MMP-9 and mulberrofuran H was chosen after docking for 20 ns MDs to give insight into the binding stability and the flexibility of the complex using Gromacs 2019. The apo protein (MMP-9 structure without ligand) was also used as the reference one. The MDs were evaluated by the values of RMSD, RMSF, Rg, percentage of hydrogen bond occupancy and hydrophobic interactions and SASA
The RMSD values of protein-ligand complexes reached stable state after 1 ns while the apo protein gained equilibrium state about nearly 6 ns, respectively (Figure 4). The variation of MMP-9 in the complex was quite large with the RMSDs from 2.0-3.0 Å compared to the first position (Figure 4). The RMSD values of ligand were found to be less than 1.0 Å after 6 ns (Figure 4). Therefore, because of ligand binding, the MMP-9 in the complex was more stable in compared with the MMP-9 apo protein.
The RMSF values showed the fluctuation of each atom throughout the simulation, especially calculated for carbon Cα of amino acids of protein. As the lack of residues from 215 to 391 in the crystal structure was the reason for the straight light (Figure 5). The RMSF values confirmed that the residues of the binding site were less fluctuable. The average RMSF values were 1.0 Å. However, residues located near the binding site (residues from 180 to 200) showed more fluctuation after ligand binding.
As can be seen from Figure 6, the Rg value of MMP-9 was nearly constant throughout 20 ns, and close to 1.5 nm in both dynamic models. The results demonstrated that MMP-9 was stable after binding to mulberrofuran H, and the ligand did not change the radius gyration of the protein.
Percentage of hydrogen bonds occupancy between protein MMP-9 (2OW0) and ligand mulberrofuran H were calculated at the different times 0, 5, 10, 15 and 20 ns (Table 6). Of the observed hydrogen bonds, mulberrofuran H showed the highest presence of important hydrogen bonds with amino acids His401 (83.07%), Pro421 (77.67%), followed by Leu188 (36.16%) and Gln402 (27.32%), similar to the interactions of the experimental complex. In addition, mulberrofuran H also formed the hydrogen bonds with Met422, Tyr420, Ala417 with high occupancy of more than 65%, showing the good interactions with S1’ pocket.
Through 20 ns simulation of molecular dynamics, the hydrophobic interactions between the benzopyran backbone of mulberrofuran H with His401 was very stable. This is an important hydrophobic interaction in the experimental complex, which determined the MMP-9 inhibitory potential of mulberrofuran H. In addition, there were also interactions between benzopyran scaffold with Leu418, Arg424 and hydrophobic interactions of the tricyclo substituent group and the amino acids Leu187 and Leu188. All the interactions were quite stable, helping the complex to bind firmly (Table 7).
Figure 7 showed that there was not much difference in SASA values of the apo protein and the protein in the complex. SASA values of MMP-9 protein fluctuated in the range of 90-95 nm2 while the MMP-9 in the complex was in the range of 95-100 nm2. This suggested that the binding of mulberrofuran H to MMP-9 did not affect the SASA values much as the folding structure of the MMP-9 protein could keep the protein in a stable state. There was also a fluctuation of SASA values in the initial 15 ns before reaching a stable state until the end of simulations.
4. DISCUSSION
The docking results on MMP-1 and MMP-9 shared many similarities with the same 4 top candidates, mulberrofuran H, mulberroside C, kuwanon S and moracin C. The common feature in the structures of these ligands was the heterocyclic groups: benzofuran (mulberrofuran H, moracin C), furochromane (mulberroside C) and chromon groups (kuwanon S) which could easy get into the S1 ‘pocket of MMP-1 and MMP-9 and interact with the pockets. The heterocyclics are not too cumbersome, so the S1 pockets of MMP-1 and MMP-9 were accessible to them, demonstrating their inhibitory potential. However, mulberrofuran H, mulberroside C and moracin C exhibited better binding on MMP-9 than MMP-1. This could be explained by the fact that the S1 ‘pocket in the MMP-9 was deeper, so the ligand could go deeper and interact with the protein better. In addition, the S1 ‘pocket in MMP-9 was narrower than MMP-1, leading to a weaker binding result for bulky structures, such as kuwanon S.
On the other hand, mulberrofuran H, mulberroside C and kuwanon S have a curved V-like configuration, having a substituted group as an anchor, which was well compatible with the shape of the binding cavity for a more stable attachment. Specifically, mulberrofuran H, with a structure of 5-benzofuran 1,3-diol was well-fitted into the pocket. However, moracin C with the isobutylene group could not push the heterocyclic group into the binding pocket, so the interactions of this compound and proteins MMP-1 and MMP-9 were weaker.
The remaining 5 compounds, morusinol, quercitin, kuwanon E, mulberrofuran Y and cudraflavone B did not have good binding affinities due to their bulky heterocyclic structure, along with the formation of endo-molecular hydrogen bonds, making them difficult to access to the S1 ‘pocket. For example, the -OH substituents on the furochromane ring of quercitin, kuwanon E, morusinol and cudraflavone B created the endolytic hydrogen bonds with cetone oxygen, increasing the cumbersome effect. Mulberrofuran Y contains the hydrocarbon dimethylocta substituent on the benzofuran group and the bulky 1,3-diol benzene group at the end of the molecule which was found not able to get into the S1’ pocket even though the structure is similar to mulberrofuran H.
In terms of interactions between investigated ligands and MMP-1 protein, most of the potential compounds mimicked the co-crystalized ligands to form the hydrogen bond with the key residue Glu219 (Table 5). Mulberrofuran H, mulberroside C and moracin C all belonged to 5-benzofuran 1,3-diol benzyl backbone, so they shared some hydrophobic interactions in the S1’ binding pocket. The chromane scaffold of the kuwanon S also created a hydrophobic bond with this cavity. The ligand mulberrofuran H generated a hydrogen bond with Pro238 and two hydrophobic interactions between its cyclohexan ring and the key residues Leu181 and His218. The mulberroside C formed hydrogen interactions with Leu181 and Ala182 based on some OH substituents of the pyranose ring. Comparing with the ligand moracin C, a hydrophobic interaction with the residue His228 of S1 pocket was generated by this ligand’s hydrocarbon methyl but-2-en substituent. The kuwanon S with two -OH phenol groups on the chromane ring created two hydrogen bonds in S1’ pocket with Ala182 and Tyr237. In addition, there were also many hydrophobic interactions between the dimethylocta substituent of kuwanon S and Leu181, Tyr210, Tyr240 (Table 5).
As for MMP-9 (PDB: 2OW0), there were not many hydrogen bonds but mainly hydrophobic interactions between top ligands and residues in the binding site (Table 5). This might be because the S1’ pocket of MMP-9 protein was possibly more hydrophobic than that of the MMP-1 protein. The ligand mulberrofuran H was stabilized by the π-contacts with Leu188, His401, His405 and His411 while the mulberroside C fitted the protein through the hydrogen bonds with Arg424 and Thr426 by the active -OH on cyclohexan, with Gly186 and Pro421 by - OH substituent on pynanoside ring, with Leu188 and Ala189 by -OH phenol substituent of the backbone, respectively. Interestingly, the best docking conformation of moracin C generated a special interaction between -OH of the benzyl-1,3 diol substituent with Zn2+ ion. Apart from many hydrophibic and hydrophobic interactions with S1’ cavity, the chromane ring of the kuwanon S also presented an important electrostatical interaction with the positive charge of Zn2+ ion (Table 5).
There were some differences of top compounds with good binding affinities on MMP-3 as the protein cavity was larger and deeper which was suitable for bulky structures (Table 5). Mulberrofurane H had the benzopyrane scaffold to be able to enter deeply insight into S1’ site, conducting π-alkyl interactions with Leu197, Val198, His224, and Leu218, so this ligand obtained the good binding affinity (-8.87 kcal.mol-1). Mulberrofuran Y (-7.92 kcal.mol-1) generated many alkyl interactions with Leu164, Ala165, Val198 and His201 due to the dimethylocta branching fitting wellto S1’ site, which could also be observed in kuwanon S (-7.64 kcal.mol-1). Moracin C (-7.50 kcal.mol-1) was different from the others because of the π-π interaction between the 5-benzopyran, 1,3-diol heterocyclic backbone with His201 (similar to the co-crystalized ligand of MMP-3 protein). Cudraflavone B (-7.44 kcal.mol-1) is a polyphenol structure with short hydrocarbon substituents, so the compound only had few hydrophobic interactions with residues in binding pocket such as Leu164, Val198, His201 and Tyr223. Morusinol (-7.02 kcal.mol-1) with chromane heterocyclic structure created many π-π interactions with His201 and Tyr223 and many other π-alkyl interactions, the hydrocarbon substituent branch created hydrophobic interactions with the trio His201, His205 and His211. Particularly with mulberroside C, a very potential ligand on two MMP-1 and MMP-9 proteins, did not yield good binding affinity on MMP-3. This could be explained by the fact that the van der Waals interaction of the ligand with MMP-3 was not as strong as with the MMP-1 and MMP-9 proteins.
In general, combinations of binding affinities and binding interactions with three targets, MMP-1, MMP-3 and MMP-9 proteins, the complex of mulberrofuran H and MMP-9 was selected for molecular dynamics simulations (MDs) to give further insight into the binding of the compound and the MMP-9.
After molecular docking, the ligand mulberrofuran H was select as the best binding ligand with the good binding affinities into three targets, MMP-1, MMP-3 and MMP-9. Through the values of RMSD, RMSF, Rg, percentage of hydrogen bond occupancy, hydrophobic interactions and SASA, the MMP-9 in the complex was more stable in compared with the MMP-9 apoprotein. Thereby, the interactions between MMP-9 with mulberrofuran H can be found to be beneficial, helping the complex to be stable.
Combination of molecular docking and molecular dynamics simulations results, there was an important hydrophobic interaction between mulberrofuran H and His401 at the active site of the MMP-9, which determined the MMP-9 inhibitory potential of mulberrofuran H. The ligand mulberrofuran H was also stabilized into the MMP-9 protein by hydrogen bond with Pro421 (high occupancy of 77.67%). In addition, there were also interactions between the benzopyran scaffold with Leu418, Arg424 and hydrophobic interactions of the tricyclo substituent group and the amino acids Leu187 and Leu188. All the interactions contributed to the stability of the complex. The results demonstrated the anti-aging potency of mulberrofurane H through the in silico study of inhibition of MMP-9 protein. Mulberrofurane H was also reported by the inhibitory activity of tyrosinase activity of Mulberrofuran H [17]. The in vitro and in vivo biological tests are required for the confirmation.
Conclusion
Through molecular docking, nine selected natural compounds from Morus alba Moraceae were investigated the prospective anti-aging agents by binding into three targets, MMP-1, MMP-3 and MMP-9. All the phytochemical compounds showed binding abilities into the targets. Of which, mulberrofuran H showed the best binding affinity on all three proteins and the complex of this compound with MMP-9 was selected for MD simulations. The values of RMSD, RMSF, hydrogen bond occupancy, hydrophobic interactions, Rg and SASA demonstrated that the mulberrofuran H and MMP-9 complex was highly stable during the period of 20 ns. An important hydrophobic interaction between mulberrofuran H and His401 was identified, which determined the MMP-9 inhibitory potential of mulberrofuran H. The ligand mulberrofuran H was also stabilized into the MMP-9 protein by hydrogen bonds with Pro421 with the high occupancy of 77.67%. These results demonstrated the inhibitory potential of mulberrofuran H on the protein MMP-9 which could be further tested for anti-aging activity.