






1. INTRODUCTION
According to Globocan statistics, stomach cancer ranked the fifth most common cancer in 2022. Currently, radiotherapy or chemotherapy need extensive treatment and often cause side effects that significantly impact the patient’s health. As a result, research and clinical applications have focused heavily on the development of new pharmaceutical substances capable of killing cancer cells in a safe manner, as well as effective delivery systems.
Stressed or damaged cells can cause cell death including apoptosis, necrosis and autophagy. Apoptosis, a caspase-mediated programmed cell death process, characterized by distinct morphological characteristics, including plasma membrane blebbing, formation of apoptosome, chromosome condensation, nuclear fragmentation and cell shrinkage. Finally, the cells are fragmented, creating apoptotic bodies, which are rapidly destroyed by macrophages without eliciting inflammatory responses [1,2]. In contrast, necrosis is defined as an uncontrolled cell death process characterized by cellular organelle swelling, plasma membrane rupture and the release of intracellular contents, leading to inflammation [3]. Apoptosis plays crucial role in numerous processes inside multicellular organisms, including development, differentiation, proliferation, homeostasis, regulation of immune function and elimination of harmful cells [2]. However, cancer cells frequently resist apoptosis, resulting in uncontrolled proliferation. As a result, inducing apoptosis in cancer cells is regarded as a safe and essential therapeutic approach. Numerous biologically active pharmaceutical substances, such as rosmarinic acid, have been demonstrated to trigger apoptosis in cancer cells and are intensively investigated as potential treatments for gastric cancer [4–6].
Rosmarinic acid (RA) is found in plants from the Boraginaceae and Lamiaceae families. It has several positive functions, including as anti-inflammatory [7], antibacterial [8], antioxidant and anticancer properties [9–12]. RA can prevent cancer by inhibiting tumor cell growth [10,12,13], arresting cell cycle [6] and cell apoptosis induction [10,12]. RA’ is a polyphenol that is poorly soluble in water and readily ionized [14]. The RA’s high acidic nature results in ineffective membrane permeability and low bioavailability [6,13,14], which is defined as the relative amount of a drug that enters the systemic circulation unaltered [15]. Bioavailability is affected by some factors including method of drug treatment and absorption [16]. Gastrointestinal fluids and gut microflora also cause structural changes in polyphenols, thereby decreasing bioavailability [5]. Several studies demonstrated that RA is significantly unstable in low acidic medium at pH 2.5 [17] or in the presence of human digestive juices [17–19]. Zoric et al. reported a great instability of pure RA in gastric phase (30.77%). The plant-extracted RA was also significantly degraded in both gastric and intestinal phases [17] due to various digestive juice components such as enzymes and bile salts [20].
The instability of RA in digestive system reduces the oral bioavailability, restricting its use in functional foods and plant-based pharmaceutical industries [5]. A delivery mechanism, such as cyclodextrin [21], phospholipid complexes [22], or nanoparticles [23,24], may be required to protect RA from alterations, thus increasing the bioavailability and sustaining antitumor efficacy of RA. Encapsulating RA in solid lipid nanoparticles protected it from degradation of gastric enzymes in acidic pH [17], retained the antioxidant activity of RA until it reaches small intestine [24]. Oral delivery of RA-loaded lipid nanoparticles only resulted in accumulation in the small intestine, where RA was progressively released and degraded [25].
As a new generation material in drug delivery system, silica nanoparticles have been most studied and developed because of their outstanding properties. Silica nanoparticles have silanol (Si-OH), siloxane (Si-O-Si) groups on their surface. These groups are next to each other and joined by hydrogen bonds, allowing silica nanoparticles easily aggregate into bigger particles [26]. Mesoporous organosilica nanoparticles are made by directly hybridized organic moieties in an inorganic silica framework using organic bridging alkoxysilanes (R’O)3Si–R–Si(OR’)3 (R as the organic radical). These nanoparticles are biodegradable because organic radicals can be decomposed under redox conditions, pH and enzymes [27]. Mesoporous organosilica nanoparticles have high biocompatibility and low bioaccumulation [28]. These nanoparticles can be loaded with large amounts of hydrophilic or hydrophobic medicines and released the action of trypsin [29] or intracellular glutathione [4,30], which break the internal bonds of particle’s structure.
In previous study, we produced new biodegradable organosilica nanoparticles - called as SiNPs - with the average size of ~50 nm and the pore volume ~1.06 mL/g. Its large surface area (498.05 m2/g) resulted in high RA adsorption capacity (580.1 mg of RA per 1 g of SiNPs). These RA-loaded particles, known as RA@SiNPs, could be absorbed by two gastrointestinal cancer cell lines and cause cytotoxicity [31]. However, the exact mechanism of their toxicity remained unknown. Since RA has the potential to induce apoptosis in some types of cancer cells [10,12], we hypothesized that the cytotoxicity of RA@SiNPs was caused by the effect of RA compound loaded in the porous particle structure and occurred via apoptosis. Therefore, this study aimed to evaluate the apoptosis-inducing potential of RA@SiNPs in AGS human gastric carcinoma cells based on several molecular and morphological changes in cells, including phosphatidylserine translocation, caspase-3 activity, DNA fragmentation and apoptotic cell morphology [1,2].
2. MATERIALS AND METHODS
AGS human gastric cancer cell line (CRL-1739) was purchased from American Tissue and Culture Collection (VA, USA). Roswell Park Memorial Institute (RPMI) 1640 medium, Fetal Bovine Serum (FBS) and Penicillin/Streptomycin were obtained from Thermo Fisher Scientific (MA, USA). Rosmarinic acid, paclitaxel (PTX), Trypsin/EDTA, FBS, phosphate buffered saline (PBS), Cell Counting Kit-8 (CCK-8), Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit and Hoechst 33342 were purchased from Sigma-Aldrich (MO, USA). Caspase-3 assay kit was purchased from Abcam (MA, USA). E.Z.N.A Tissue DNA Kit was purchased from Omega Bio-Tek (VT, USA). GelRed® Prestain Plus 6X DNA Loading Dye was purchased from Biotium (CA, USA). Hyperladder 1 kb was obtained from Bioline (London, UK). The SiNPs and RA@SiNPs organosilica nanoparticles were received from Center for Innovative Materials and Architectures (Ho Chi Minh City, Vietnam) [31].
The study included the evaluation of the effects of RA-loaded nanoparticles on cell growth by using cytotoxicity assays, and the demonstration of the cytotoxic mechanism involving apoptosis by various assays. First, healthy cells were incubated with the reagents for 24 hours and the medium was renewed. After an additional 48 hours of culture, the assays were performed (Fig. 1).
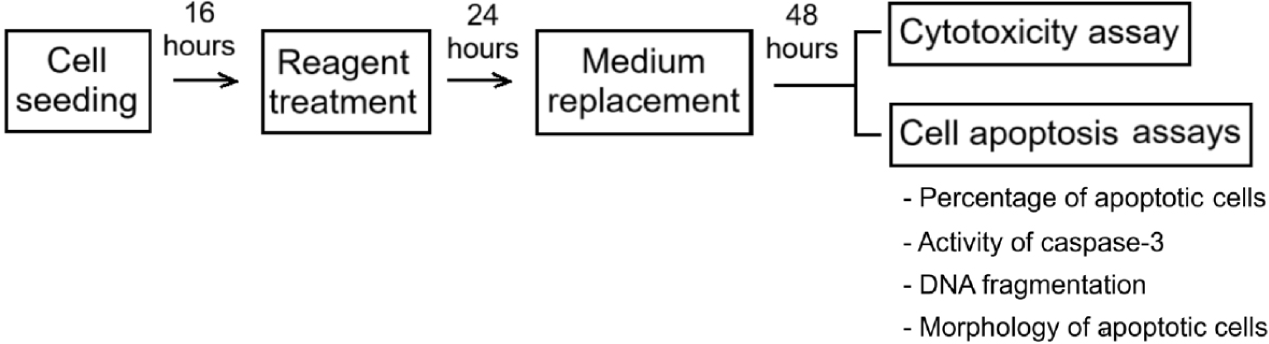
SiNPs and RA@SiNPs (2.4 mg/mL) were dissolved in sterile distilled water to create stock solutions. RA was completely dissolved in sterile distilled water at a concentration of 1 mg/mL and PTX stock (50 mg/mL) was produced in DMSO, following the manufacturer’s instructions. For testing, stock solutions were diluted with culture medium at the highest concentration and then gradually diluted to lower concentration solutions.
The completed RPMI 1640 medium was prepared with supplementation of 10% FBS and 1% Penicillin/Streptomycin. AGS cells were cultured in full medium at 37℃ in a humidified environment containing with 5% CO2. For testing, a quantity of cells was sown in each well of a microplate for 16 hours to ensure that cells were in proper growth phase before doing the assay. Then, cells were treated with different concentrations of reagents (RA@SiNPs, SiNPs, RA) or with a positive control PTX, a commonly used anticancer drug that is effective in cell cycle arrest and induces cell death [32]. Negative controls were untreated cells to ensure that any changes identified in the test group were due to the influence of the reagent, and not due to other factors, thereby underlying comparison with the test group. After 24 hours of reagent incubation, the wells were gently spun several times and discarded old medium through a vacuum suction pump. Cells were rinsed with 1X PBS and reconsituted with freshly prepared media. Cellular analyses were then done 48 hours later [4,31].
Cell viability was evaluated using the CCK-8 assay [31]. Briefly, 3×103 cells were seeded in each well of a 96-well plate, treated with reagents (RA@SiNPs, SiNPs, RA, and PTX), or left untreated (negative control), as previously described. N=3 wells/group. The RA@SiNPs concentrations were 30, 60, 120 and 240 μg/mL, corresponding to 17.5, 35, 70 and 140 μg/mL of RA contained in the particles, respectively [31]. The SiNPs concentrations of were 30, 60, 120 and 240 μg/mL, while RA concentrations of 17.5, 35, 70 and 140 μg/mL. PTX (2, 20 μg/mL) was used as the positive control. The final volume of solution in each well was 100 μL. To analyze, 10 μL of CCK-8 solution was added to each well of plate. After 2 hours of incubation, absorbance was measured at 450 nm using microplate reader (Varioskan Lux, Thermo Fisher Scientific, MA, USA). The experiment was repeated three times.
Cytotoxicity was assessed based on the calculation of cell viability ratio (CVR) by the following formula:
where ODs: optical density of sample,
ODo: optical density of negative control.
The percentage of apoptotic and necrotic cells can be determined using Annexin V-FITC/PI test, as per the manufacturer’s recommendations. Briefly, 5×105 cells were seeded in each well of a 6-well plate and treated with optimal doses of RA@SiNPs (240 μg/mL of SiNPs loaded 140 μg/mL of RA) or RA (140 μg/mL) as mentioned previously. Untreated cells served as negative controls. N=2 wells/group. Cell suspensions were collected into a conical tube. The remaining adhering cells were collected with 1 mL trypsin-EDTA at 37℃ for 5 minutes and transferred into the same tube. After centrifugation at 1,200 rounds per minute for 5 minutes, cell pellet was collected and resuspended in 500 μL of binding buffer. Cells were stained with 5 μL of Annexin V-FITC and counter-stained with 10 µL of PI for 10 minutes in darkness. Analysis was immediately performed using a flow cytometry system (FACSCanto II, BD Biosciences, CA, USA), with excitation wavelength at 488 nm. Emission filters for Annexin (green) and PI (red) were 533 nm and 585 nm, respectively. A total event of 10,000 cells per sample was collected. The experiment was repeated three times.
The enzyme activity was measured using colorimetric Caspase-3 Assay Kit according to the manufacturer’s instructions. To begin, 5×105 cells were seeded in each well of a 6-well plate and incubated with RA@SiNPs (240 μg/mL), SiNPs (240 μg/mL) or RA (140 μg/mL), as mentioned previously. Untreated cells served as negative controls. N=3 wells per group. All suspending and adherent cells were harvested. After centrifugation, the cell pellet was lysed with 1X lysis buffer. The total amount of protein was measured at 280 nm-wavelength. 50 μg of protein was added into each well of a 96-well plate and incubated with 50 μL of 2X reaction buffer/DTT and 200 μM of DEVD-p-nitroaniline substrate at 37℃ for 90 minutes. Caspase-3 activity was measured by quantifying the p-nitroaniline light emission using a microplate reader (Varioskan Lux, Thermo Fisher Scientific, MA, USA) at 400 nm. The experiment was repeated three times.
An amount of 5×105 cells were seeded in each well of a 6-well plate and tested with 240 μg/mL of RA@SiNPs, 140 μg/mL of RA or without reagent (as negative control), as mentioned previously. N=2 wells/group. All suspension and adherent cells were collected. Cell pellets were collected after centrifugation at 1,200 rounds per minute for 5 minutes. Genomic DNA was isolated using an E.Z.N.A Tissue DNA Kit according to the manufacturer’s instructions. Briefly, cell pellet was resuspended in 200 μL of pre-chilled 1X PBS and mixed thoroughly with 25 μL Proteinase K Solution. Protein was discarded through a binding column. DNA was precipitated with absolute ethanol. DNA quantity was measured using a NanoDrop spectrophotometer (NanoDrop-2000, Thermo Fisher Scientific, MA, USA). DNA samples were stained with GelRed® Prestain Plus 6X DNA Loading Dye and immediately analyzed on a 1.5% agarose gel at 100 V for 30 minutes. The experiment was repeated three times.
An amount of 104 cells/well was cultured in an 8-well chamber slide, incubated with 240 μg/mL of RA@SiNPs or without reagent (as negative control). N=2 wells/group. After 24 hours, the chamber was removed, and the slide was fixed with 4% formaldehyde solution for 15 minutes. Cells were stained with 10% Giemsa/PBS solution or with 1 µg/mL pre-chilled Hoechst 33342/PBS solution for 10 minutes. After being washed several times with 1X PBS, cells were directly reviewed and photographed under a fluorescent microscope (BX53, Olympus, MA, USA). The experiment was repeated three times.
All experiments were triplicated, and the results were presented as means±SD. Calibration curves were constructed using a linear regression model (Microsoft Excel, Microsoft, 2018). The Kruskal-Wallis test was employed with GraphPad Prism 8.4.2 to compare group means when variances were unequal or when data distributions between two groups deviated from normality. A p-value of less than 0.05 (*) or less than 0.01 (**) was considered statistically significant. The Shapiro-Wilk test was used to assess the normality of data distribution, whereas Bartlett’s test was employed to navigate variance homogeneity among groups. The Kruskal-Wallis test was specifically applied to compare differences between samples and negative controls in the cytotoxicity assay, variations among different cell types in the Annexin V-FITC/PI apoptosis assay, and differences in optical density (OD) values in the Caspase-3 activity assay. For post-hoc pairwise comparisons following the Kruskal-Wallis test, Dunnett’s test was used.
3. RESULTS
The cytotoxicity was investigated using CCK-8 assay, in which the OD value was converted into CVR percentage. Analytical results revealed that the toxicity of RA@SiNPs, SiNPs and RA solutions was proportional to their concentrations (Fig. 2). The dose of 140 μg/mL of RA resulted in the lowest cell survival rate in both RA@SiNP-treated cells and RA-treated cells, which were 54.31±9.16% and 43.60±7.80%, respectively (Table 1). In particular, toxicity of SiNPs in AGS gastric cancer cells was not detected at particle concentrations lower than 120 μg/mL. The CVR achieved at the highest concentration of SiNPs (240 μg/mL) was 86.65±1.81%. As a positive control, low concentration of PTX could inhibit the survival of AGS cells better than RA and RA@SiNPs.
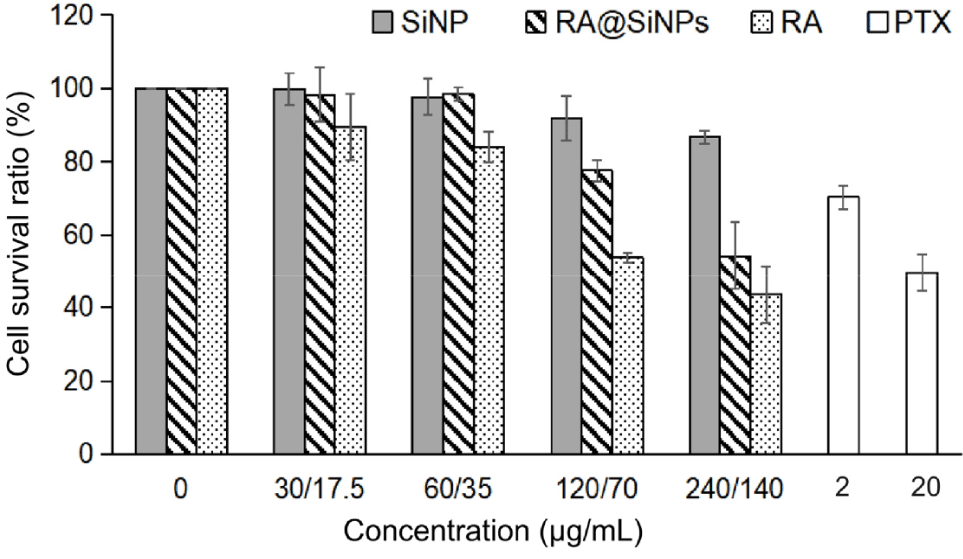
AGS cells were incubated with different doses of RA@SiNPs, SiNPs, RA, and PTX, or without reagent (0 μg/mL, considered as negative control) for 24 hours. Each sample was tested on three wells. After 48 hours of additional culture in fresh medium, the cytotoxic was assessed by the CCK-8. The OD measurement was performed at 450 nm and converted into the cell viability ratio. Data were expressed as mean±SD of three independent experiments. All p-values were more than 0.05. CVR, cell viability ratio; RA, rosmarinic acid; PTX, paclitaxel; AGS, adenocarcinoma; CCK-8, Cell Counting Kit-8; OD, optical density, NA, not applicable.
Cell death can be occured through a various mechanisms, including apoptosis, necrosis and autophagy. This study aimed to assess the apoptosis-inducing ability of RA@SiNPs in AGS human gastric carcinoma cells based on several molecular events and morphological changes in cells during apoptosis.
Firstly, early-stage cell apoptosis was characterized by the translocation of negatively charged phosphatidylserine from the inner layer to the outer layer of plasma membrane [2] where it facilitated specific membrane binding to the Annexin V, which is conjugated with a FITC fluorescent dye to identify apoptotic cells in flow cytometry. Cell apoptosis was identified in both RA@SiNPs-treated and RA-treated groups and was undetectable in the negative control group (Fig. 3). The total proportion of apoptotic cells at both early and late stages in RA@SiNPs-treated and RA-treated groups were approximately 43% and 35% of total cells, respectively. In particular, the rates of necrotic cells in both RA@SiNPs-treated and RA-treated cell groups were rather low, accounting for 6.90±0.14% and 0.55±0.21%, respectively (Table 2).
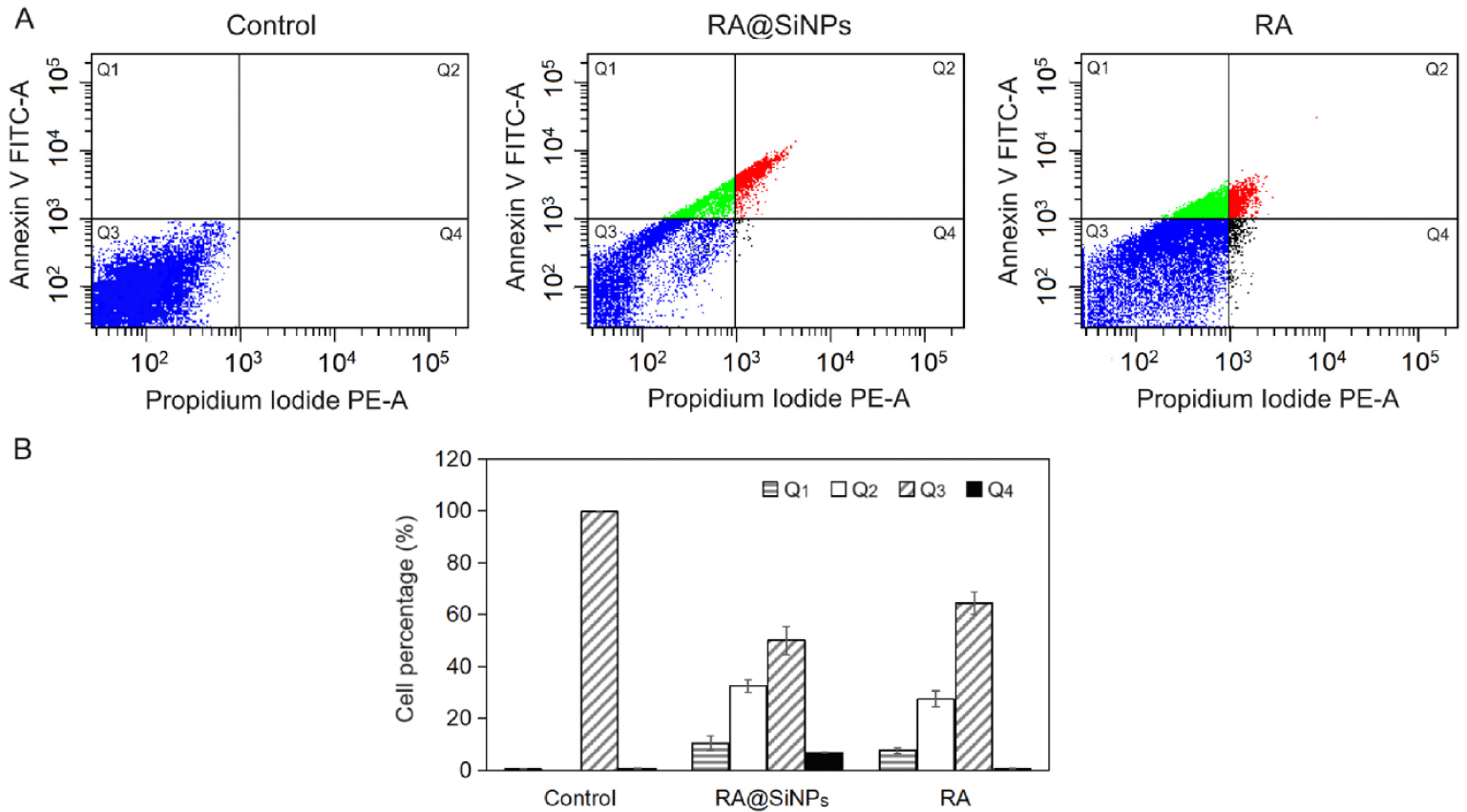
AGS cells were incubated with 140 μg/mL of RA or RA@SiNPs for 24 hours. Untreated cells were the negative control. Each group was performed in two wells. Cells were stained with an Annexin V-FITC/propidium iodide kit and detected by a flow cytometry system. Data were expressed as mean±SD of three independent experiments. All p-values were more than 0.05.
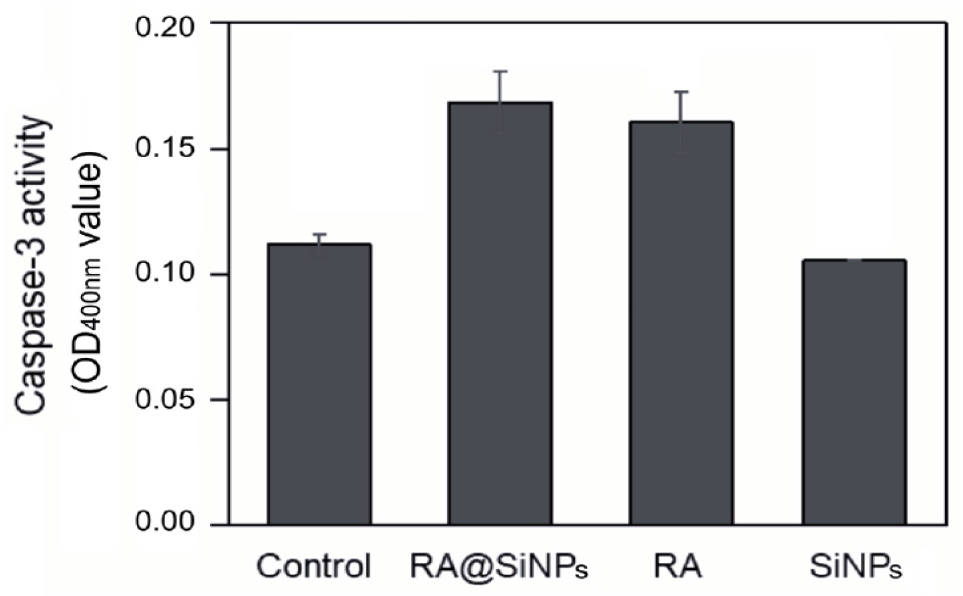
A hallmark of the late phase of apoptosis is the chromosomal DNA degradation by endonucleases which are activated by caspase-3, a cysteine protease [2]. In this study, the activity of caspase-3 in apoptotic cells could be assessed by the cleavage of the DEVD-p-nitroaniline substrate resulting in light emission at a wavelength of 400 nm. Caspase-3 activity increased in AGS cells treated with RA@SiNPs and with RA. In contrast, this enzymatic activity in cells incubated with SiNPs was as low as that in the negative control (Fig. 4).
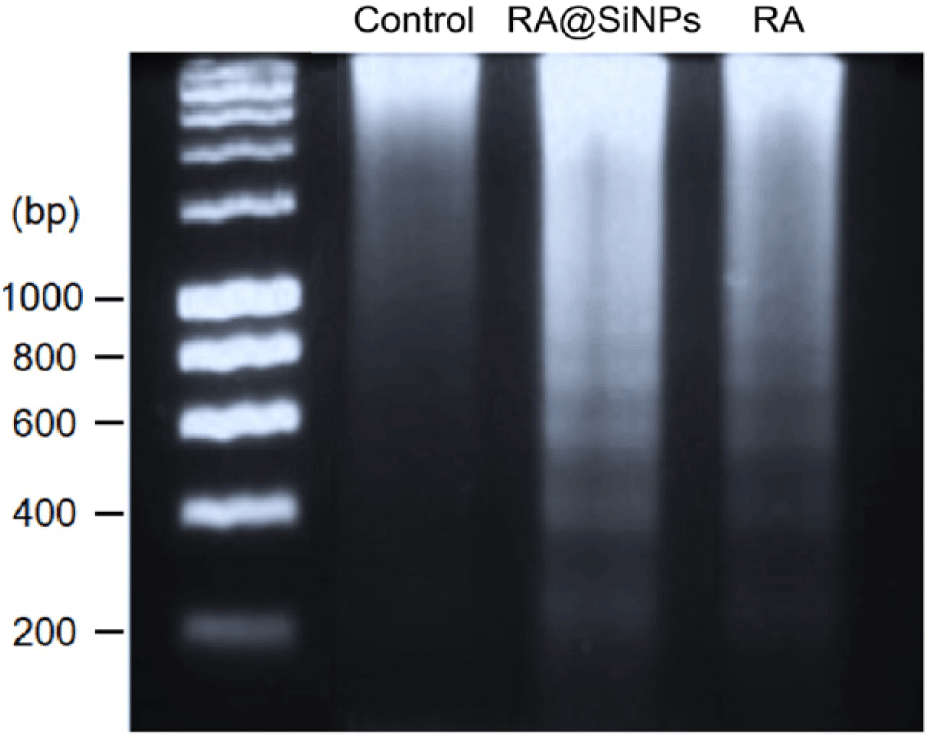
Afterward, caspase-3 activated for the release of caspase- activated DNase which initiated the chromosomal DNA cleavage into oligonucleosomal fragments in approximate multiples of 180–200 base pairs size, as observed in the results of DNA electrophoresis of RA@SiNPs-treated and RA-treated cell groups, but not in the negative control (Fig. 5).
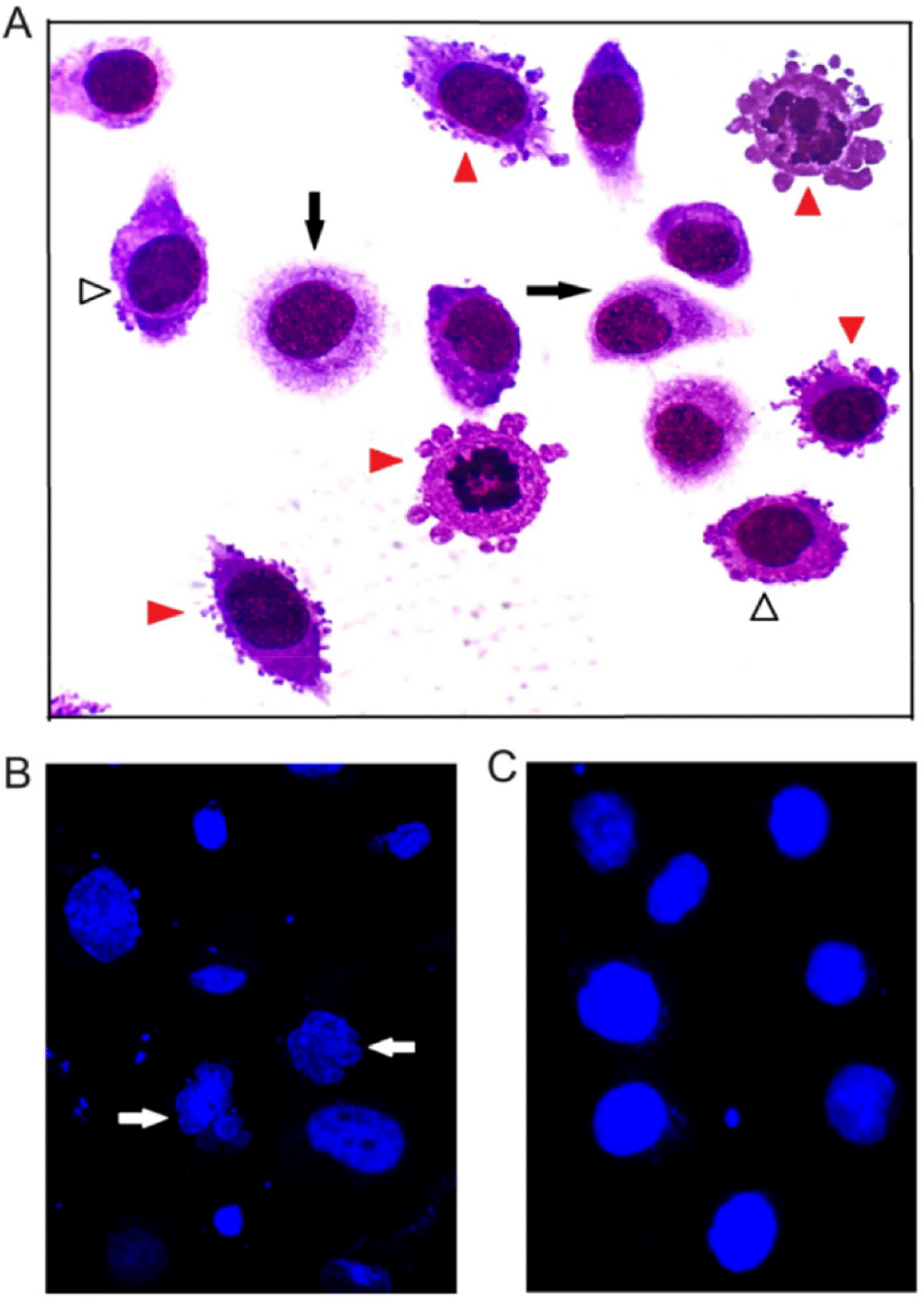
DNA degradation begins when nuclear condensation is complete, and it is accompanied by major changes of cell morphology in the late stage of apoptosis [2,33]. These modifications can be investigated using microscopic techniques. AGS cells treated with RA@SiNPs and stained with Giemsa’s solution (Fig. 6A) presented nuclei shrinkage and chromatin condensation (white arrowheads), and plasma membrane blebbing (red arrowheads). Furthermore, fluorescent analysis of Hoechst-stained nuclei revealed significant chromatin compaction and nuclear fragmentation (Fig. 6B, white arrows) compared with normal nuclear morphology (Fig. 6C).
4. DISCUSSION
The natural compound RA possesses beneficial medicinal properties such as antioxidant, antibacterial [28], anti-inflammatory [7] and anticancer activities [9–12]. However, its low oral bioavailability prohibits its beneficial properties from being employed as medications [6,13,34]. Our previous findings showed that encapsulating RA in mesoporous organosilica nanoparticles not only protected it from acidic environments but also inhibited the survival of gastrointestinal cancer cells [31], but the mechanism of cell death induced by RA@SiNPs required further investigation. This study demonstrated that using a drug delivery system like RA@SiNPs caused toxicity to AGS human gastric carcinoma cells in dose-dependent manner (Fig. 2), and this cytotoxicity was promoted by apoptosis (Figs. 3–6). SiNPs have low toxicity to both cancer cells and normal cells [4]. It is probable that apoptosis was triggered by RA released from the structure of RA@SiNP [31,35] via intracellular BAX/BCL-2 pathway [10,36] or EGFR/Akt/NF-κB pathway [36].
RA has limited bioavailability due to its poor water solubility and membrane permeability [6,13,34]. Therefore, encapsulation of RA with nanoparticles is a solution to improve cellular absorption of RA [23,24]. However, the cytotoxic effect of RA released from RA@SiNPs was reported to be less effective than the RA that directly absorbed into AGS cells when administered at the same concentration (Fig. 2). We assumed that this toxicity was influenced by the quantity of nanoparticles that have been taken up into cells within 24 hours of treatment. Although confocal microscopic analysis indicated evidence of intracellular SiNPs in prior studies, cellular uptake efficiency was not established [31]. In addition, effective cytotoxicity also depends on the efficiency of RA release from the pore structure of nanoparticles, because of the destruction of disulfide/tetrasulfide bridges in the organosilica nanospheres [4]. Redox conditions, intracellular pH [8,37] and intracellular antioxidant glutathione tripeptide were the factors that caused this characteristic degradation [4,30,38]. Cytotoxic efficiency also depended on the concentration of reagent and the exposure time [9,10]. To achieve 140 μg/mL of RA, an amount of 240 μg/mL of nanoparticles were required, because the loading capacity of RA@SiNPs was around 580.1 mg of RA per 1 g of SiNPs [31]. Increasing the concentration of particles could obscure the cell layer, making it difficult to observe the cell shape and disturbing the OD values. The prior experimental technique called for a 24 hour incubation time for the reagent [4,31]. Increasing incubation time of RA@SiNPs may increase cell uptake efficiency, but continuous cell proliferation over that period can increase cell density which affects OD measurement in untreated or SiNPs-treated groups. Improvements in the structure of SiNPs to enhance RA loading efficiency and intracellular absorption are very important to ensure the proper release of herbal compounds, resulting in enhanced cytotoxicity and apoptosis in AGS cancer cells. Therefore, we believe that several factors such as the cell absorption efficiency of RA@SiNPs, the RA release efficiency from nanoparticle structure, and the cell incubation time of RA@SiNPs are limitations that need further research.
RA was considered non-toxic for normal cell lines [31,39,40]. Its selective cytotoxicity may reduce common side effects of conventional cancer treatment. In this study, cytotoxic effects of RA@SiNPs and RA were compared to PTX, a commonly used anticancer drug that is effective in cell cycle arrest and cell death [32]. While only 20 μg/mL of PTX was used to achieve 49.70±5.23% of viable cells, approximately 70–140 μg/mL of RA was required to achieve equivalent CVR (Table 1). It is possible that the lipophilic nature of PTX enables its rapid and abundant absorption through cell membranes, resulting in substantial accumulation in cancer cell lines. Because of the rapid and dramatic effects on cell cycle arrest and cell death, the efficacy of PTX in cancer therapy has been authenticated [41,42]. Meanwhile, RA is a poorly aqueous soluble polyphenol [14]. The ineffective membrane permeability [6] and decreased stability in low pH of environment [17] can make RA less effective.
Even though PTX had better cell inhibition that involved in apoptosis pathway, it also caused autophagy [42,43] and necrosis [42], which releases intracellular materials and activates undesirable inflammatory responses that can lead to further tissue damage [3]. Meanwhile, apoptosis is a process of physiological cell death. Individual cells undergo apoptosis, causing unique nuclear and cytoplasmic alterations. Finally, the apoptotic cell is rapidly digested by macrophages, without stimulation of inflammation [2]. Therefore, encouraging the body’s natural mechanism of apoptosis to kill cancer cells is a promising strategy to cancer therapy today [12]. The findings of this study confirmed the ability of RA@SiNPs to kill AGS gastric cancer cells through apoptosis without involving necrosis. The usage of phytomedicines loaded with nanomaterials like RA@SiNPs may reduce unwanted inflammation in cancer patients.
5. CONCLUSION
Cancer cells can multiply uncontrolled and avoid planned cell death. Although rosmarinic acid has the potential to decrease cell survival by activating the apoptosis pathway in some cancer cell types, its low bioavailability is a barrier for in vivo medication delivery. By encapsulating rosmarinic acid with degradable mesoporous organosilica nanoparticles, this herbal compound was able to be delivered into AGS gastric cancer cells and inhibited cell survival. The capacity of RA@SiNPs to cause apoptosis in cancer cells increases their usefulness in safe anticancer therapy.