







1. INTRODUCTION
Hypertension is a common chronic medical condition that can cause cardiovascular complications such as cerebrovascular accident, heart failure, coronary heart disease and myocardial infraction. Nowadays, ramipril is commonly used to treat heart failure and high blood pressure. Ramipril aids in the prevention of heart attacks, future strokes, and kidney problems. According to USP 43 [1] and BP 2020 [2], there were 4 specified impurities that need to be controlled in ramipril substance and finished products. Among ramipril specified impurities, ramipril methyl ester (impurity A) is important to be used and tested in ramipril pharmaceutical substances and corresponding finished products. Currently, there is no reference standard of impurity A in the reference substance bank system of the National Institute of Drug Quality Control and the Institute of Drug Quality Control in Ho Chi Minh City. This related compound is costly with limited accessibility, which affects implementation of this impurity test. There have been no published studies on chemical synthesis of impurity A so far. Therefore, this study was conducted with the aim of synthesis and establishment of reference standard impurity A. Finally, the assay of impurities in available ramipril pharmaceutical products was conducted by HPLC method, using the established impurity A reference standard.
2. MATERIALS AND METHOD
Ramipril substance: Batch number 5654-19-122M, purity 99.85% on anhydrous basis, loss on drying 0.1%, expiry date 04/2022, obtained from ZHEJIANG HUAHAI Pharmaceutical Co. Ltd. Ramipril reference standard: Lot number LRAC3757, purity 99.8% on the basis, loss on drying 0.01%, expiry date 08/2023, obtained from Sigma - Aldrich RTC.
Melting point was measured on DSC Q20 - TA Instruments. IR spectrum was recorded on Shimadzu IRAffinity - 1S; Mass spectrum was recorded on Varian Ultramass 700; 1H-NMR and 13C-NMR spectra were recorded on Bruker Advance II spectrometers. Liquid chromatography was performed on system Alliance 2695 XE (Water), equipped with a photodiode array (PDA) detector.
HPLC grade acetonitrile (ACN) and HPLC grade methanol (MeOH) were obtained from Fisher and Merck. HPLC grade trifluoroacetic acid (TFA) and HPLC grade triethylamine (TEA) were obtained from Prolabo. All other chemicals reagents and solvents were acquired from commercial sources and used without further treatment. Samples of Ramipril tablets were purchased from a market in Ho Chi Minh city in April 2021.
Ramipril impurity A was synthesized by transesterification reaction of ramipril and methanol. Potassium hydroxide was used as the catalyst for the transesterification [3] (Figure 1).
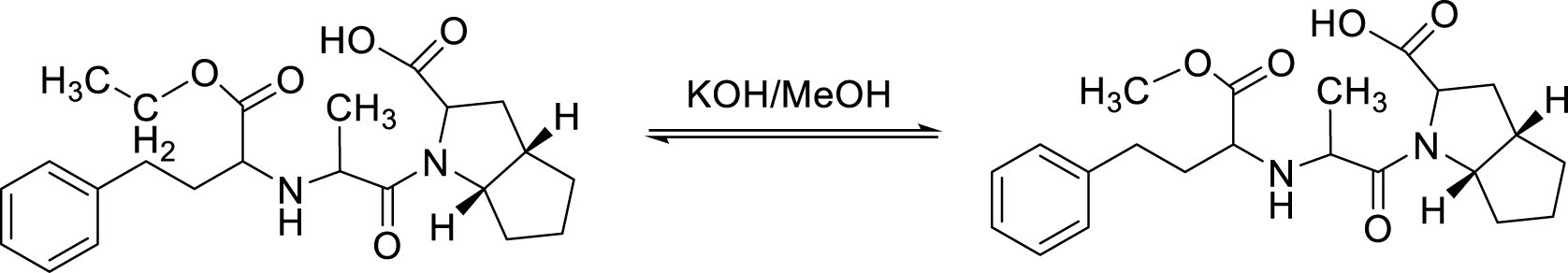
The synthesized product was precipitated, washed with water, and refined by recrystallization in organic solvent. The purity of the refined product was checked by thin layer chromatography (TLC) in 3 different polarities mobile phase and melting point test. TLC was performed using F254 silica gel thin sheet (10 x 2 cm); detected at wavelength 254 nm. The refined product was diluted in methanol solvent at a concentration of 1 mg/mL. Mobile phases used in TLC: n-hexane - ethyl acetate (1: 3); n-hexane - MeOH (1: 1); n-butanol - acid acetic - water (8: 1.5: 0.5).
The synthesized and refined product chemical structure was elucidated from its IR, MS, and NMR spectroscopic data.
Purity of synthesized impurity A was determined by HPLC-PDA based on peak area normalization method. 10 μL of solution impurity A was injected into column Eurospher II C18 (250 x 4.6 mm; 5 μm) at 40 °C. The PDA detector operated at wavelength of 210 nm. Mobile phase was a mixture of solvents I and II maintained at a flow rate of 1 mL/min. Solvent I was ACN, and solvent II was solution TEA 0.01% with additional TFA to get pH 4. The mobile phase was a mixture of solvent I and solvent II in gradient program. Solvent I remained at 10% linearly increased to 45% for 10 min and kept for 5 min, then continuously increased from 45 to 90% for 5 min and returned to 10% over 5 min before the next injection. Total chromatographic runtime was 25.0 min.
The HPLC-PDA purity determination procedure was validated in accordance with International Council on Harmonization (ICH) Guidelines [4].
This HPLC procedure was also used to determine the assigned value of chromatography purity when setting up the reference standard impurity A at 3 laboratories of the Institute of Drug Quality Control in Ho Chi Minh City.
Impurity A was evaluated by characters: appearance, solubility, melting point, loss on drying, identified by IR, MS and NMR spectroscopic methods and purity by HPLC.
After the bottling process, samples were taken for evaluation of the homogeneity of bottling process at random, the number of vials sampled followed the formula Vn+1, where N is the total number of vials. The determination of purity of impurity A in each vial was carried out according to the developed and validated HPLC method, in which each vial was measured twice. The assessment of the homogeneity of the bottling process was based on the guidance of ISO 17034:2016 [5]. The homogeneity of the bottling process was accepted if the coefficient of variation (CV) between calculated purity of impurity A from taken vials did not exceed 1%.
After the evaluation of the homogeneity of the bottling process, the interlaboratory vial homogeneity was determined. The purity of impurity A in sampled vials was determined in three independent laboratories. The laboratories selected to submit samples should meet requirements of ISO/IEC 17025:2017 [6]. Each laboratory received six random sample vials alongside the related documentation. Prior to analysis, each laboratory was examined for the suitability of its chromatographic system. The suitability was accepted if impurity A peak asymmetry factor (As) was in the range of 0.8 - 1.5 and CV between 6 replicated measurements did not exceed 2%. After that, the determination process of impurity A purity was conducted on 6 received samples in each laboratory, twice for each sample. The laboratories selected to analyze samples were the Department of Standardization & Reference Materials (Laboratory 1), the Department of Cosmetic Testing (Laboratory 2), the Department of Research and Development (Laboratory 3) of the Institute of Drug Quality Control in Ho Chi Minh city. The evaluation of interlaboratory vial homogeneity was performed by one-way analysis of variance (ANOVA).
The determination of the assigned value in the impurity test of impurity A and measurement uncertainty were carried out according to the guidelines of ISO 13528:2015 [7], based on 18 results of the determination of impurity A purity in 3 laboratories.
Impurity A after establishment of reference standard and reference standard impurity D from our previous report [8] were implemented in the impurity test of finished ramipril products. HPLC-PDA procedure was performed using column Gemini C18 (250 x 4.6 mm; 5 μm) at column temperature of 65 °C and with PDA detector operating at wavelength of 210 nm. Mobile phase was a mixture of solvent A and solvent B in gradient program at a flow rate of 1.0 mL/min and injection volume was 10 μl. Solvent A was prepared by dissolving 2 g of sodium perchlorate in a mixture of 0.5 mL of TEA and 800 mL of water, adjusted with phosphoric acid to a pH of 3.6 before adding 200 mL of ACN. Solvent B was prepared by dissolving 2 g of sodium perchlorate in a mixture of 0.5 mL of TEA and 300 mL of water, adjusted with phosphoric acid to a pH of 2.6 before adding 700 mL of ACN. Over 0-6 min, solvent A remained at 70% and linearly decreased from 70 to 30% for 24 min and kept for 10 min, then returned to 70% over 5 min, remained for 5 min before the next injection. Total chromatographic runtime was 50.0 min.
After testing system suitability, the procedure was used to determine of impurities in ramipril products samples.
Sample solution nominal concentration was 1 mg/mL of ramipril was prepared as follows: A total of 20 tablets of ramipril pharmaceutical product containing ramipril were weighed and then finely powdered with a mortar and pestle. After that, an amount of the powder equivalent to 10.0 mg of ramipril was transferred to a 10 ml volumetric flask with 6 ml of solution A, sonicated for 30 min with intermittent shaking to dissolve and then was diluted to volume with solution A and filtered through a 0.45 μm pore size membrane filter.
System suitability solution of 50 μg/mL was prepared by dissolving requisite amount each of standard reference ramipril, standard reference impurity A, and standard reference impurity D in solution B.
Standard solution of 5 μg/mL ramipril was prepared by dissolving requisite amount of standard reference ramipril in solution B.
Sensitivity solution of 1 μg/mL ramipril was prepared by diluting of standard solution five times in solution B.
System suitability was performed by injecting system suitability solution, standard solution, and sensitivity solution into the chromatographic system. System suitability was accepted if in the chromatogram of system suitability solution, resolution (Rs) was not less than 3.0 between peaks of ramipril impurity A and ramipril; in 6 chromatograms of standard solution, CV of value peaks area of ramipril did not exceed 5.0%; in chromatogram of sensitivity solution, signal-to-noise ratio (S/N) was not less than 10.
Based on analysis of standard solution and sample solution, the calculated percentage of each impurity in the portion of tablets taken:
Ru: peak response of each impurity from the sample solution; Rs: peak response of ramipril from the standard solution; Cs: concentration of ramipril reference standard in the standard solution (mg/mL); Cu: nominal concentration of ramipril in the sample solution (mg/mL); F: relative response factor [1]. Acceptance criteria for test impurity were showed in Table 1.
3. RESULTS
3.0 g (7.40 mmol) of ramipril substance, 0.3 g of sodium hydroxide and 30 ml of methanol were put in a round- bottomed flask. The flask was heated and stirred at 85 °C for 90 minutes. After that, the reaction mixture was cooled to 35 °C and then was concentrated under reduced pressure. 15 ml of water was added to the residual mass and stirred, then hydrochloric acid was slowly added to adjust the pH to 4. The resultant precipitation was filtered, washed with water, dried at 70 °C to a constant weight and finally recrystallized from isopropanol 3 times to yield 1.08 g (2.9 mmol) of white, odorless powder. The efficiency of the whole process was about 39.2%.
The obtained product melting point was measured three times. The measured range of melting temperature was 136.0 - 137.5 °C, which was consistent with the melting point of impurity A in reference [9]. The results of TLC on three solvent systems with different polarities indicated that the product only gave a single trace in chromatograms. The product was preliminarily concluded to be purified.
IR spectrum of synthesized product showed characteristic absorption peaks of functional groups, including (Vmax, cm-1): 3268 (NH); 2953 (CHsp3); 1745, 1649 (C=O acid); 1195 (CO ester). Positive ESI-MS of product revealed an [M+H]+ at m/z = 403.2234 and negative ESI-MS of product revealed an [M-H]- at m/z = 401.2030 (calculated accurate mass = 402.2155), both of which corresponded to molecular weight of 402.48 with molecular formula C22H30N2O5.
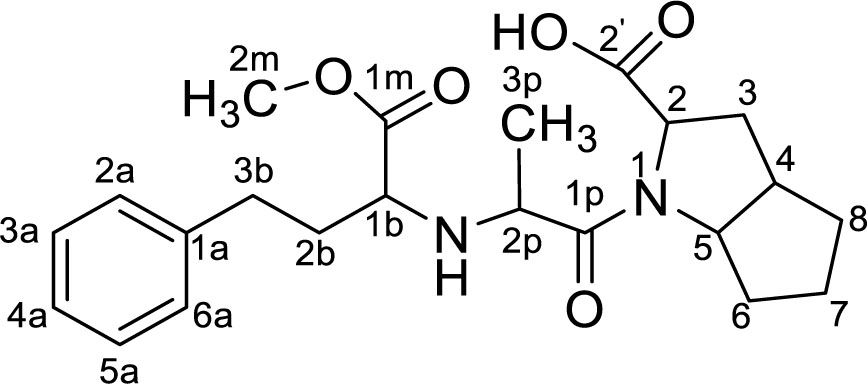
NMR data: 1H-NMR (500 MHz, CD3OD, δ ppm): 7.30 (t, J=7.6 Hz, 2H, H3a, H5a); 7.27 - 7.21 (m, 2H, H2a, H6a); 7.23 - 7.16 (m, 1H, H4a); 4.25 - 4.07 (m, 4H, H2p, H2, H5); 3.70 (s, 3H, H2m); 2.92 - 2.75 (m, 2H, H1b); 2.71 (dt, J=14.0 Hz, 1 H, H3b); 2.41 - 2.30 (m, 2H, H3), 1.85 (ddd, J=10.3; 7.5; 4.6 Hz, 2H, H2b); 1.76 - 1.53 (m, 4H, H6, H7, H8); 1.37 (d, J=7.0 Hz, 3H, H3p) (Figure 2).
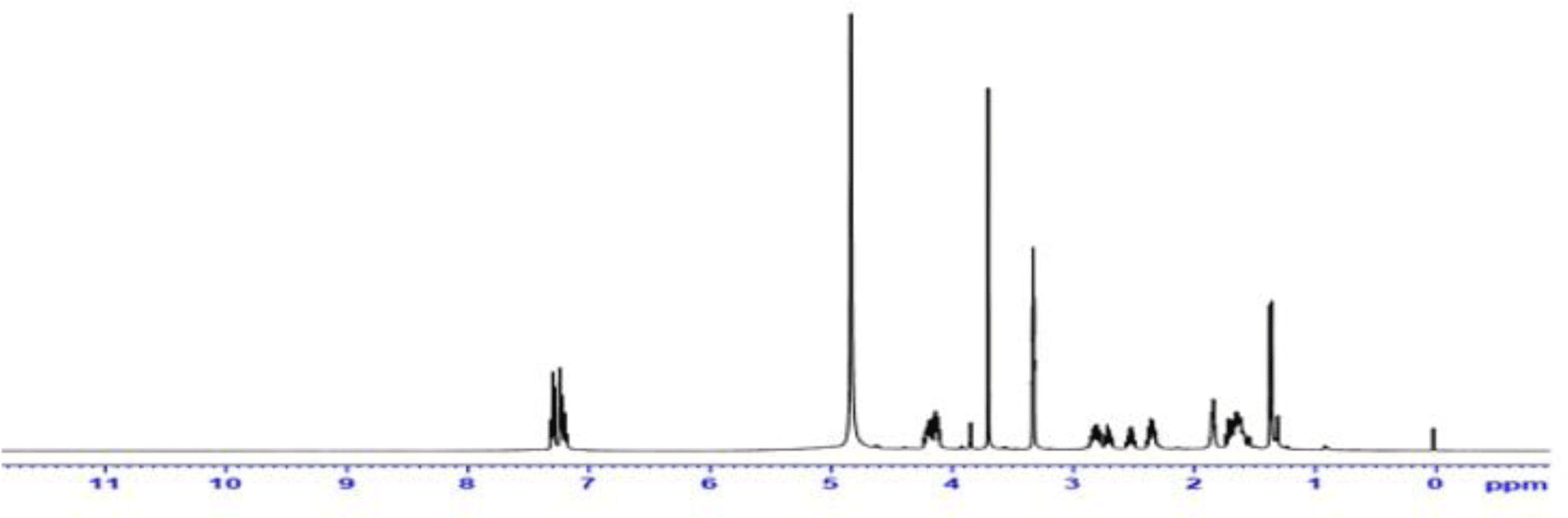
13C-NMR (125 MHz, CD3OD, δ ppm): 172.45 (C1m); 170.49 (C1p); 168.68 (C2); 142.41 (Cu); 129.61(C2a, C6a); 129.41(C3a, C5a); 127.24 (C4a); 64.53 (C5); 61.25 (C2p); 58.51 (C1b); 58.04 (C2); 52.82 (C2m); 42.36 (C4); 34.55 (Cab); 33.49 (C6); 32.74 (C8); 32.12 (C2b); 31.06 (C7); 24.57 (C3); 16.20 (C3p) (Figure 3).
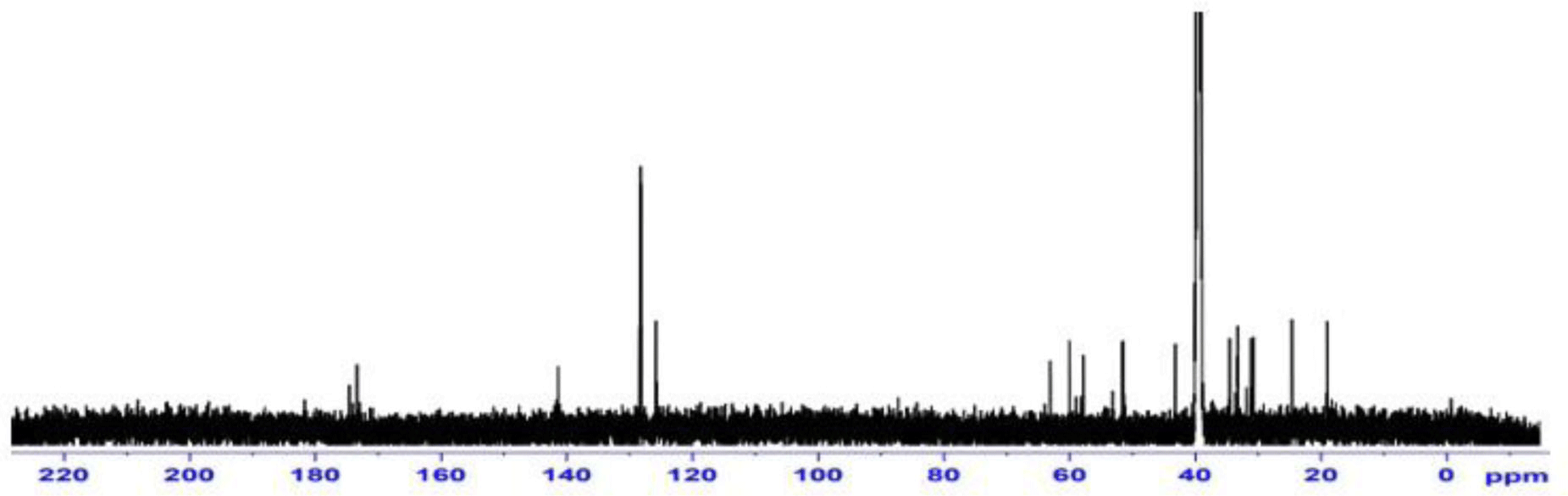
From the obtained spectral results and comparison NMR spectral data with NMR spectral data of Ramipril in literature [10, 11], it can be concluded that the synthesized and purified product is impurity A (ramipril methyl ester), molecular formula: C22H30N2O5, M = 402.48 (Figure 4).
System suitability was determined by repeating injection of solution impurity A with a concentration of 500 μg/mL 6 times. The results were showed in Table 2. Typical chromatogram of impurity A was showed in Figure 5.
| Parameters | tR (min) | S(μV.s) | As | N |
|---|---|---|---|---|
| Average | 13.79 | 10025595 | 1.0 | 3503 |
| CV(%) | 0.06 | 0.31 | 0 | 0.43 |
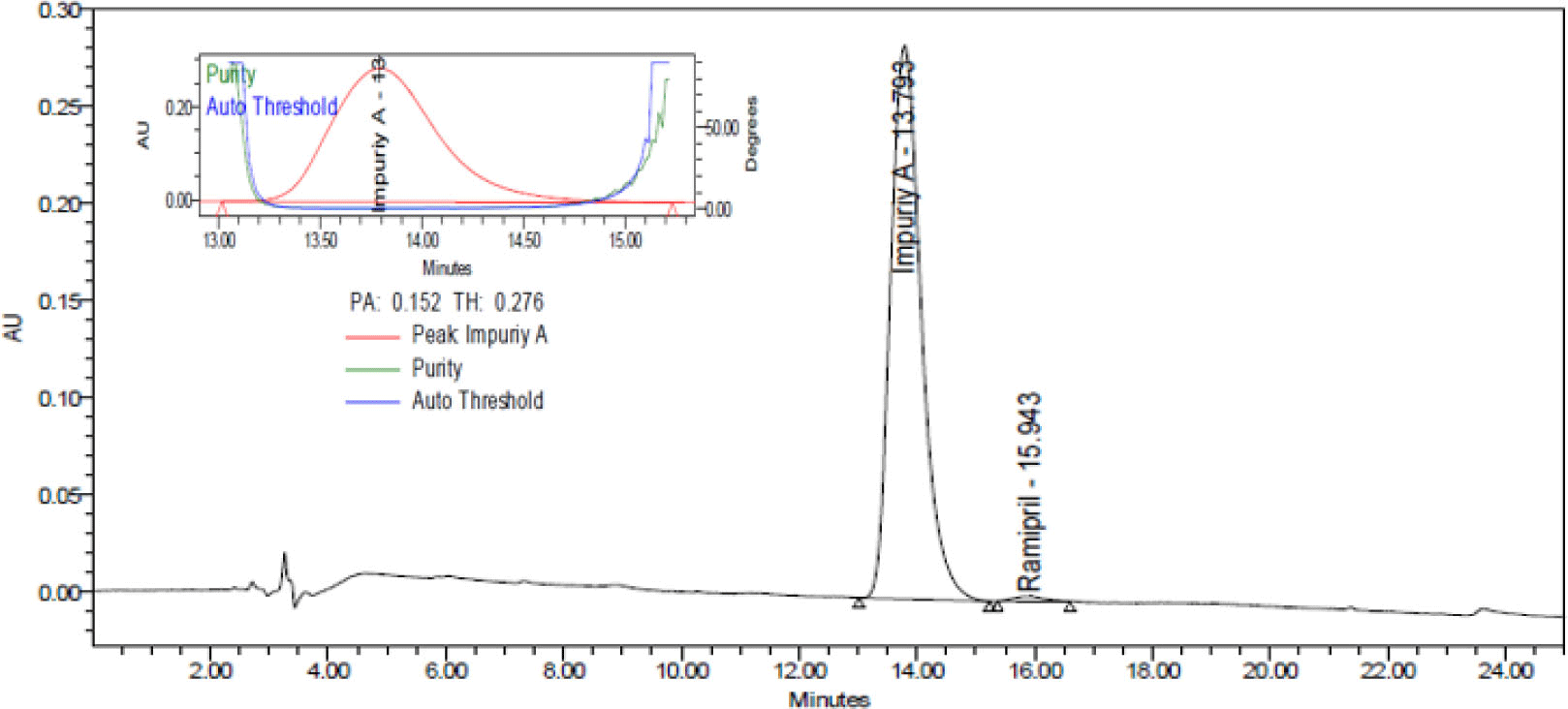
The CV of peak areas from 6 repeated injections of samples was less than 2.0%, the asymmetry factor (As) was between range 0.8 - 1.5 and the number of theoretical plates (N) more 2000. Therefore, the procedure was suitable for analysis.
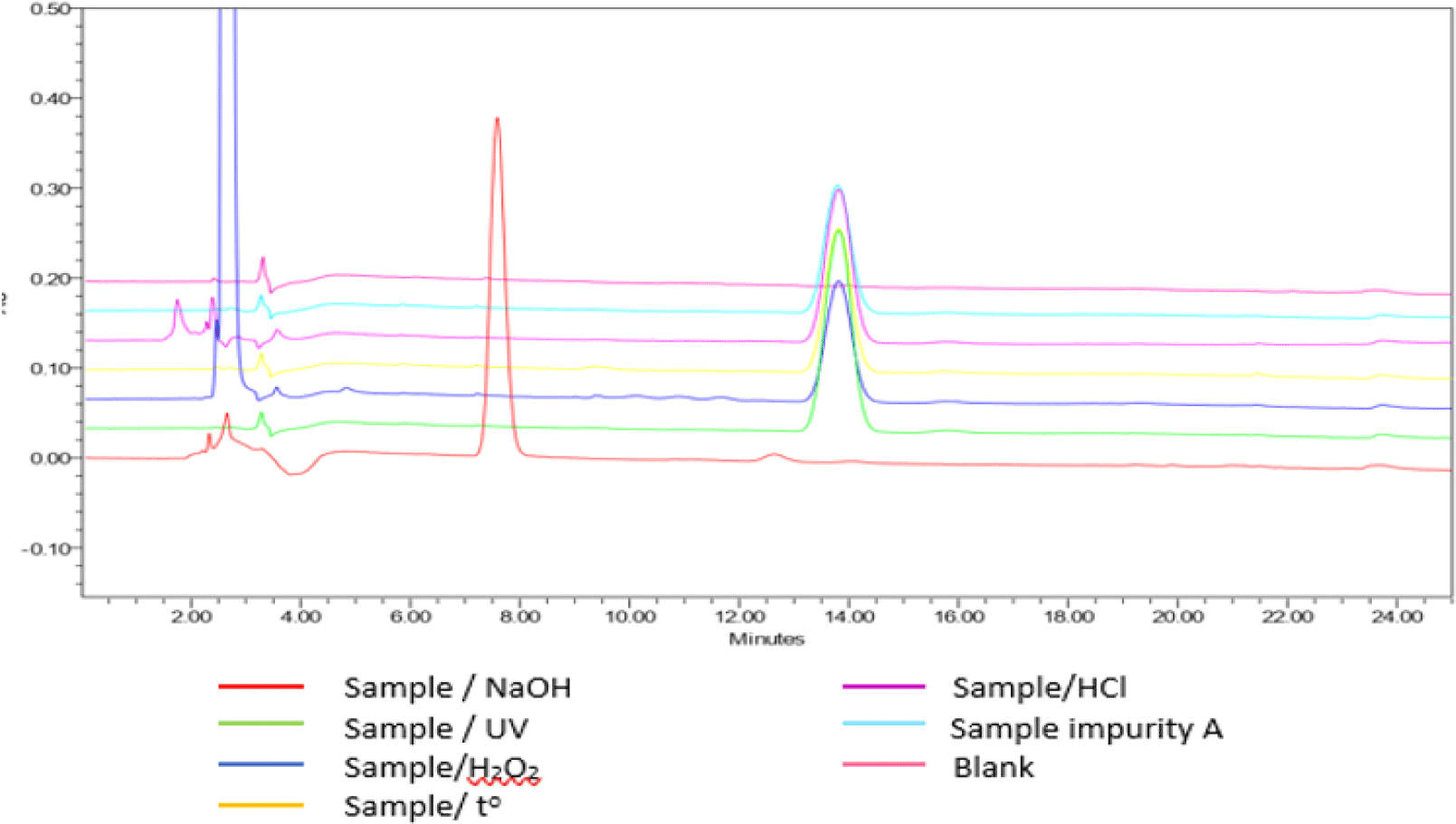
The results of selectivity testing (see Figure 6) indicated that the blank chromatogram had no peak with a retention time equivalent to that of impurity A in the sample chromatogram. Chromatogram of degraded samples, treated by 0.1 M hydrochloric acid; 0.1 M sodium hydroxide; 3% peroxide; dried at 80 °C for 24 hours and irradiated by UV 254 nm for 8 hours showed peaks of degradation products and these peaks were completely separated from those of impurity A. The peaks of impurity A in the chromatograms of the samples had satisfactory peaks purity. Thus, the procedure for determining purity of impurity A was highly selective.
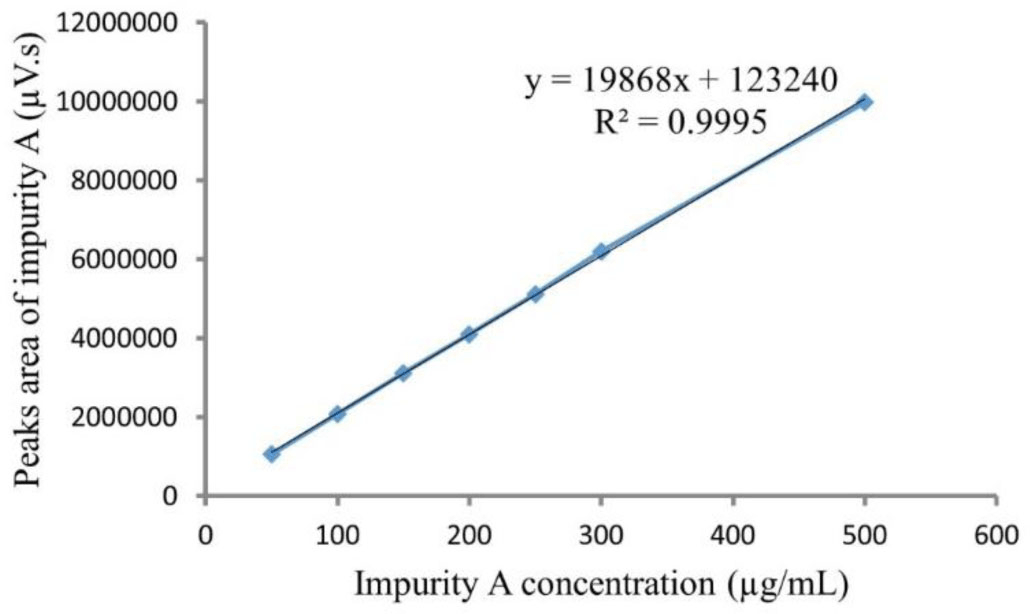
Calibration curves of impurity A ranged from 50 to 500 μg/mL. The typical linear regression equation between impurity A concentration (μg/mL) and its peak area was y = 19868x +123240 with the mean value of correlation coefficient square (R2) for impurity A calibration curve more than 0.995 (see Figure 7).
The intra-day precision levels were 99.01%, CV = 0.16% (n=6). The inter-day precision levels were 99.08%, CV = 0.15% (n=12). All inter- and intra-day assays were summarized in Table 3.
| Found mean purity | CV (%) | |
|---|---|---|
| Intra-day precision (n = 6) | 99.01 | 0.16 |
| Inter-day precision (n = 12) | 99.08 | 0.15 |
Table 4 showed results of impurity A assessment. With purity of over 98%, impurity A was eligible to be established as a reference substance.
| Characters | Method | Criteria |
|---|---|---|
| Apperance Solubility |
White, odorless powder/ Freely soluble in methanol; sparingly soluble in isopropanol |
|
| Melting point | DSC | 136–137.5°C |
| Identity | IR | IR: vmax cm-1: 3268 (NH); 2953 (CHsp3); 1745, 1649 (C=O acid); 1195 (C-O ester) |
| MS | m/z = 403.22 [M+H]+, m/z = 401.20 [M-H]-, molecular formula C22H30N2O5 | |
| NMR | 1H-NMR and 13C-NMR data are suitable with figures 2 and 3 | |
| Loss on drying | Drying at 70 °C for 3 hours | ≤0.1% |
| Related impurities | HPLC | ≤2.0% |
| Purity | HPLC | ≥98.0% on the basis |
300 mg of impurity A was packed into 30 vials of 10 mg each and 18 vials were randomly collected using Excel software for evaluation of vial homogeneity and interlaboratory vial homogeneity. The results of determining the suitability of the system at three laboratories met the requirements of the asymmetry factor of the peak impurity A and CV of impurity A peak areas. Based on the results of 6 analysis for vial homogeneity in Laboratory 1, the packed vials had uniform content, stable and suitable packaging conditions (Table 5).
| Vials ID | Purity of impurity A (%) |
|---|---|
| 2 | 98.95 |
| 13 | 98.98 |
| 4 | 98.95 |
| 19 | 98.92 |
| 23 | 99.06 |
| 11 | 99.08 |
| average | 98.99 |
| CV (%) | 0.07 |
The received results of impurity A purity between the 3 laboratories were not statistically significant, therefore analytical procedure was highly reproducible and the purity of impurity A was independent of the participating laboratory. The result of the interlaboratory vial homogeneity evaluation was shown in Table 6.
The results of determining the assigned value of impurity A were presented in Table 7.
The results suggested there was no change in the value of s* (=0.073) after the third iteration, then x* was 99.01%. Therefore, the assigned value of impurity A was 99.01% and uncertainty measurement u = 1.25s*/√p = 1.25s*/√18 = 0.0215.
Therefore, impurity A is eligible for registration of national standards with its purity of over 98% on the basis. The standard vials were labeled and attached to the test certification. Long-term storage of standard vials was at 2 - 8 °C and protected from light.
In the chromatogram of system suitability solution, Rs between peaks of ramipril impurity A and ramipril was more than 3 (see Figure 8). In chromatograms from 6 repeated injections of standard solution, CV of peak areas of ramipril was 0.32%. The ratio S/N in chromatogram of sensitivity solution more than 10. The procedure met suitability requirements.
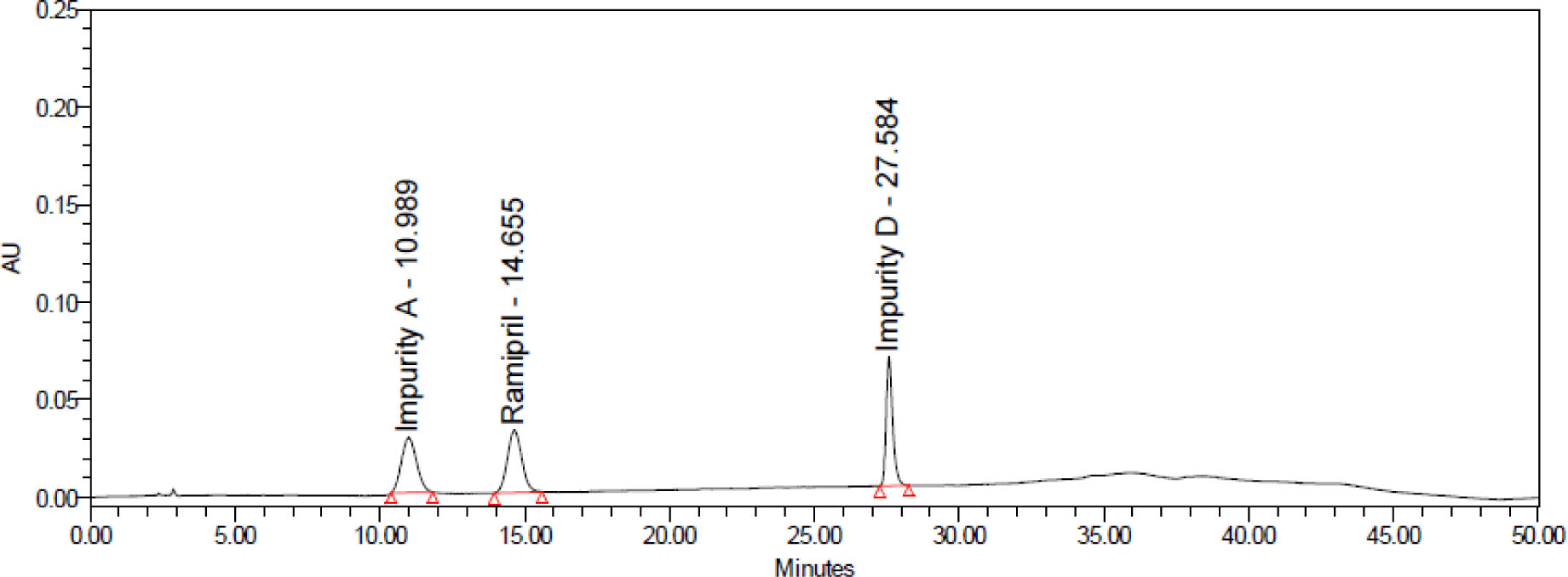
Each drug sample was analyzed twice. Chromatograms of samples was presented in Figure 9. The contents of impurities determined in 3 ramipril drug samples were shown in Table 8.
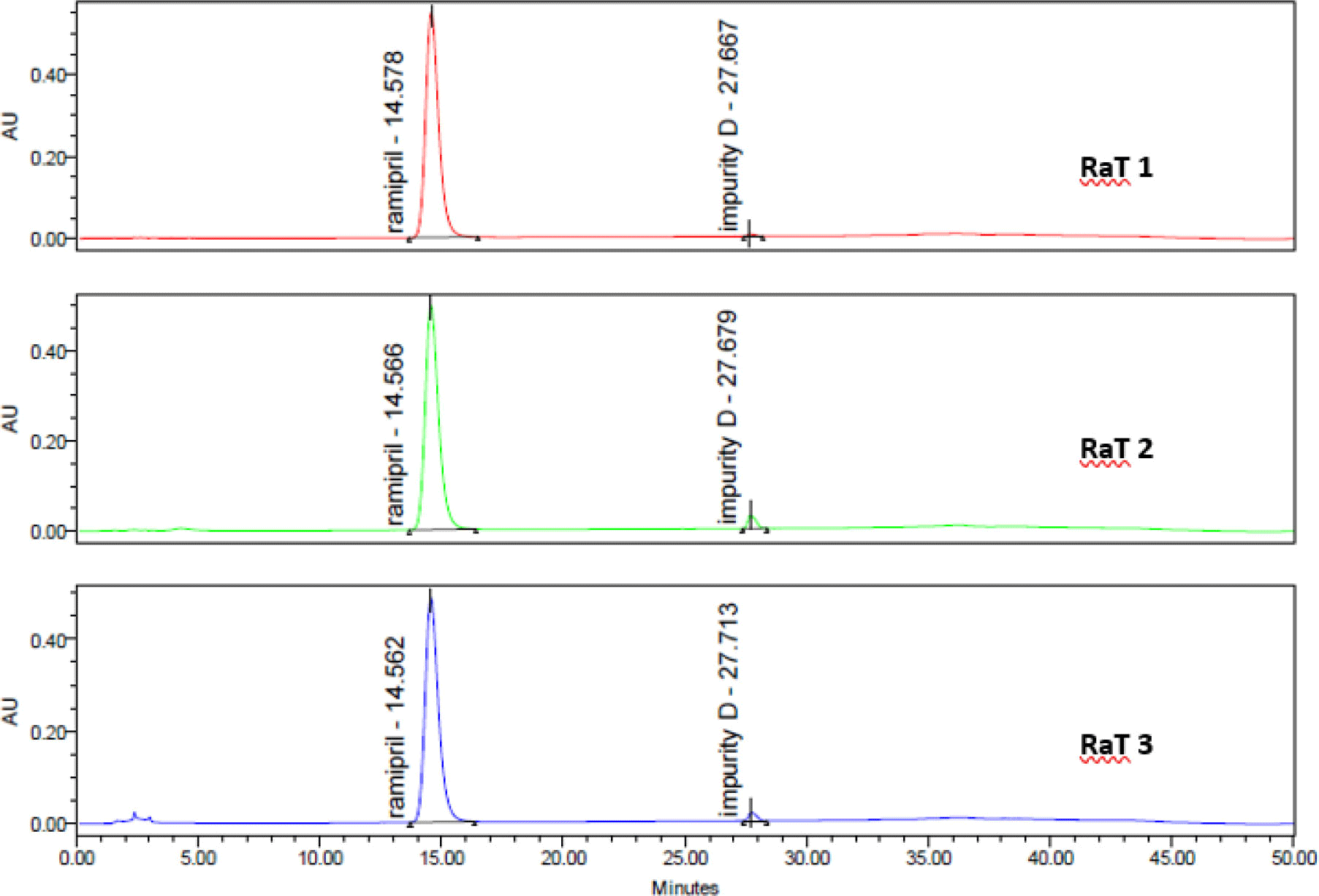
As shown in the results, the content of related impurities in the preparations were within the acceptance limits of USP 43. The impurities A, B, C were not detected in all 3 samples.
4. DISCUSSION
Transesterification is a type of nucleophilic substitution reaction. CH3O- alkoxide nucleophile was created from the reaction between methanol and potassium hydroxide. Transesterification is a reversible reaction in which ester can be hydrolyzed, especially in alkalic media. Therefore, determining the ratio of ramipril – MeOH – KOH and the temperature of reaction were important to make the reaction shift toward products and increase the reaction efficiency. Experiments indicated that the efficiency was maximized at the ratio of 1 g ramipril – 0.1 g KOH – 10 ml MeOH and temperature reaction of 85 °C. Since C2H5O- is also a nucleophile that can attack the electrophilic center, long reaction time may cause the reverse action to occur, thus reducing the efficiency considerably. From experiments, the optimal efficiency was obtained at 90 minutes of reaction time.
The reaction mechanism of impurity A synthesis was depicted in Figure 10.
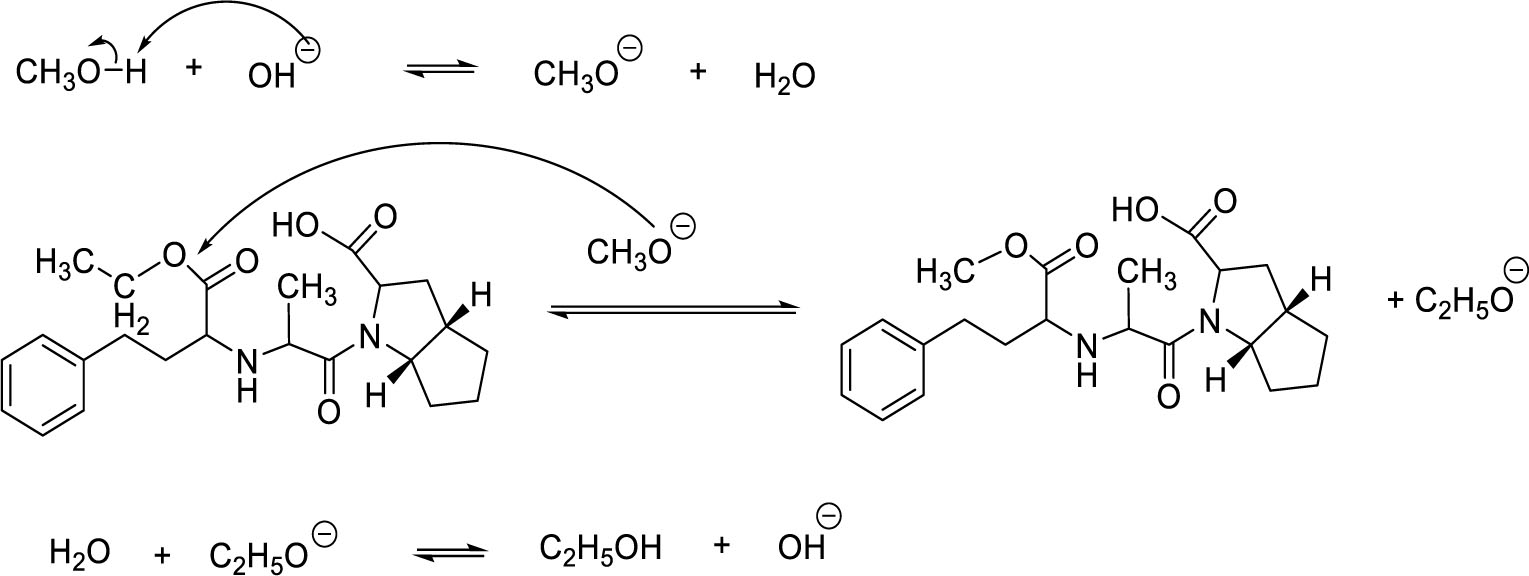
This study also determined the optimal pH condition for precipitation of synthesized product. The highest yield of impurity A was obtained at pH of 4. Recrystallizing with isopropanol increased the purity of the refined product, whereas using n-butanol and ethanol created other impurities and reduced content of crystallized product.
1HNMR and 13CNMR spectral data of impurity A were not found in literatures but comparing 1HNMR and 13CNMR spectral data of synthesized product with 1HNMR and 13CNMR spectral data of ramipril [9, 10] showed similarities. The ramipril showed additional signals of a CH2 group compared to the synthesized product. The synthesized product (impurity A, C22H30N2O5) is the methyl ester, whereas ramipril (C23H32N2O5) is the ethyl ester of ramipril acid. All received spectral data of synthesized product were completely suitable to structure of impurity A. Therefore, it can be concluded that the synthesized product is impurity A.
As in reference materials, analysis procedure of ramipril and related impurities was conducted by HPLC method with mobile phase containing sodium perchlorate. Although the perchlorate ion is effective in separating substances, it has a negative effect on the chromatographic system and column. Therefore, in the procedure of determining purity of impurity A, TFA with similar effect was used in place of perchlorate to reduce the adverse effect. As a result, in the chromatograms, peak of impurity A was completely separated from that of ramipril, with satisfactory asymmetry factor and reduced chromatographic time.
HPLC procedure for impurities test in Ramipril tablets was taken from USP 43 but with chromatographic column C18 (250 x 4.6 mm; 5 μm) instead of C18 (250 x 4.0 mm; 3 μm). However, results of checking suitability of implemented procedure were still satisfied. Column C18 (250 x 4.6 mm; 5 μm) was common in most laboratories of Vietnam. Therefore, the above chromatographic column and the impurities testing procedure in USP 43 can be used to assess of impurities in ramipril finished products. The results of impurities assay indicated that the content of related impurities in the tested preparations were within the allowable limits of USP 43.
Conclusion
Impurity A of ramipril had been successfully synthesized and standardized as a reference substance with a purity of above 98%, meeting the requirements for use in assays of related impurities. The assay related impurities of ramipril procedure according to USP 43 has been applied to check for impurities in some ramipril products on the market.